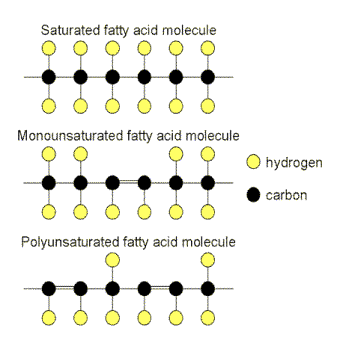
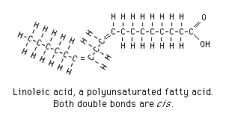
|
Essays by Malcolm
Kendrick, MD 2004 ATKINS
AND THE FIRST LAW OF THERMODYNAMICS STATINS FOR CHILDREN – THIS IS MADNESS WHY THE ATKINS DIET IS HEALTHY LIQUID DRANO-LIKE SYNTHETIC “GOOD CHOLESTEROL” PLAQUE BUSTER! IT’S SCIENCE, BUT NOT AS WE KNOW IT TELEOANALYSIS — OR WHEN I FINALLY REALIZED THAT I HAD FALLEN DOWN THE RABBIT HOLE SO, WHAT DOES CAUSE HEART DISEASE? TYPE ll DIABETES ISN’T A DISEASE? IS HEART DISEASE ALL DUE TO BLOOD CLOTS? OKAY, DO YOU KNOW WHAT A "FAT" IS? WHAT ON EARTH IS A LIPOPROTEIN? HIGH
BLOOD PRESSURE: IT’S A SYMPTOM, NOT A DISEASE, STUPID! TREATING HIGH BLOOD PRESSURE: WHAT CAN YOU BELIEVE? THE JOY OF HYPERTENSION TRIALS THE NEW HYPERTENSION GUIDELINES IDIOTIC
THINKING IN MEDICINE -
C-REACTIVE PROTEIN. THE
DEATH OF THE REFERENCE An
Example From Heart Disease Research And Yet Another Reason Why The
Scientific Machine Is Close To Meltdown
by
Malcolm Kendrick MD
*** It is often said of statistics that
scientists use them like a drunk uses a lamp-post, for support rather than
illumination. I suppose the whole point of a scientific
reference is that it is used, primarily, to provide support, so I can't
really complain about the lack of illumination. However, I can complain
about the fact that the abuse of references has led to the point where I
have found that, increasingly, I can't be sure if what I read is true
anymore. As some readers will know I have a particular
fascination with heart disease. Some would call it an obsession, but I
prefer the word, fascination. It's that old Y chromosome thing. Anyway what
I have found, as I have researched away, is that a number of bold statements
of fact, even those referenced to the gunnels, when exposed to a bit of
scrutiny, crumble to dust. For example, at one stage I was interested
why women, in most Western Countries, suffer a much lower rate of heart
disease than men - at least until about sixty five or seventy. Women usually
have very similar risk factors to men therefore, according to conventional
wisdom, there should be little difference in deaths from heart disease. I suspect I know what you are thinking. Women
are protected against heart disease by their sex hormones. This was what I
used to think as well as well. Probably because I had seen this fact stated
so many times, in so many papers, that I had been brainwashed into believing
that it was true. This belief was reinforced by the ‘knowledge' that
female protection seemed to disappear not long after the menopause, so I
didn't really think to question it. Nor, it seemed, did anyone else. Just to
quote from one study, which accurately reflected mainstream thinking a few
years back: 'A
protective effect of estrogen is the most obvious reason for the substantial
and consistent favored status of women vs. men with regard to coronary heart
disease.' Barrett-Connor E Atherosclerosis Dec 1995 However,
I started to find out a number of facts about women and heart disease that
made me begin to question things. Firstly, it was clear that if you gave
female sex hormones to men, their rate of CHD increased dramatically. Which
doesn't prove anything for sure, but it does give pause for thought. After
all, there is no reason why a chemical that protects women shouldn't also
protect men. Then
I found that there were some populations where younger women suffered the
same rate of heart disease as men, such as women in Brazil. In addition to
this, the Framingham study showed that, when women developed type II
diabetes their relative protection against heart disease disappeared, even
at a young age. Diabetes doesn't wipe out sex hormones, so where did the
protection go? As
this type of information began to pile up, I started to suspect that women
may not be protected by sex hormones after all. And so I was galvanised into
action and set out to track down the study, or studies, that had been
carried out proving that women are protected by sex hormones. In
effect I went on the quest to track down the original source of this
hypothesis. Which I knew from previous experience can be very hard work. Just
because it states in a recent scientific paper that female sex hormones are
protective, doesn't mean that the authors did a study. They are usually just
quoting from another paper, which has quoted from another paper, which
quoted from another paper, which quoted from… sometimes it seems
ad-infinitum. Trying to track backwards in time to find that very first
study from which all other studies sprang is not the work of a single day. You
can, it sometimes seems, find yourself late at night reading manuscripts
with illuminated script, penned by Monks in the early fourteenth century.
‘ And it has been rightly noted by the good physics of this borough
that the clutching disease of the breast is more common in the male than the
female, and that the protection of fair lady folk be due to the most strange
substance of eastrogenne found in the most fecund of ye womanne .' Well,
not quite, but I once chased down a reference linking heart disease to
impotence and found the source reference from a study in Germany in 1928 (In
German). Other
times I have found that there is no source study at all. The whole thing has
sprung to life from thin air. On bad days I sometimes think that references
are no more than the scientific equivalent of rumour and gossip. ‘Ooooh,
you'll never guess what, sex hormones protect against heart disease.' ‘Who
told you that.' ‘The
Mayo clinic did a study, I think. Or at least that's what Harvard said.' ‘Really?'
‘And
I read it in the Lancet too.' ‘Oh
well, it must be true. I'll have to go off and tell all my students.' And
then they'll go off and tell all their students. Some of whom will write
papers starting with the comment. ‘It is known that women are protected
against heart disease by their sex hormones.' Then other people will use
their papers as references, then… After
a while this rumour reaches the point where it becomes an unquestioned truth
because so many people have said it so many times, and it has been written
in hundreds of papers. In addition, experts stand up at conferences and
re-affirm it. Which means that when you write ‘Sex hormones protect women
against CHD,' you are then able to support this with hundreds and hundreds
of references from major journals? But
in this case, where was that original study, the source reference? What was
it all based on? Well, gentle readers, it was based on nothing at all.
Because there never was an original study. Or if there was, it is so damned
well hidden that I have never able to find it (he said covering himself from
the inevitable pedant who will no doubt triumphantly unfurl the 1936 trial
done in Serbia-Montenegro). All
I could find were papers referring to other papers that referred to other
papers. Yes, it is true that a number of studies were done which showed that
estrogen raised HDL levels and reduced LDL levels and protected the
endothelium, and all sorts of other ‘test-tube' effects. But in the end,
if you wanted to prove that female sex hormones protect against CHD, there
are only two direct ways to do it. 1.
Remove sex hormones from younger women, and see if the rate of heart
disease goes up 2.
Add sex hormones to women who no longer produce them, and see if the
rate of heart disease goes down. Had
anyone done this? No. Actually
I lie, the correct answer is yes. In 1963, a study was done in which women
who had hysterectomies were matched with women who had hysterectomies with
removal of both ovaries (no sex hormones). And the result of this study was…..?
As
you might have guessed, the result of this study was that there was no
difference in the rate of heart disease between the two groups. ‘We
found no difference in the prevalence of Coronary Heart Disease in the
oopherectomised (both ovaries removed) and hysterectomised (no ovaries
removed) women.' Ritterband et al Gonadal function and the development
of CHD. Circulation 1963 (2) pp 237 – 51 Okay,
it's only one study, but it was totally negative, and no-one had ever done a
study that was positive. Yet, despite a complete lack of evidence, the sex
hormones theory had built to the point where millions of women around the
world were being prescribed HRT in order to prevent heart disease. All this,
resting on a piece of pure speculation that only became ‘fact' through the
process of endless repetition and cross-referencing. And when, finally, researchers decided it was time to see if HRT did actually protect against HRT, what did they find? Well, a few large studies were done, and if I may quote from the New England Journal of Medicine on the matter: ‘Estrogen
plus progestin does not confer cardiac protection and may increase the risk
of CHD among generally healthy postmenopausal women, especially during the
first year after the initiation of hormone use. This treatment should not be
prescribed for the prevention of cardiovascular disease.' Manson
et al NEJM Aug 2003 Now
then, at this point, some of you may have picked up on the gentle irony that
I am using references to support my argument that references are a load of
rubbish. But of course, references used properly, are a good thing. (I mean
the way I use them, or course) However,
references are very much a double edged sword, or perhaps a bazooka. In
the wrong hands they can do far more harm than good. And in the, essentially,
unchecked system that we now have, one careless reference can end up taking
on a life of its own. It gets stuck in the medical information ‘machine'
replicating itself like some malevolent computer virus, gradually infecting
all data and turning it into useless mush. I
don't know what the answer to this problem is. Wipe the whole database clean
and start again? Set up a system to hunt down and check all references, and
remove those that are found to be wrong. Then remove all references to those
references. The mind boggles at the size of that task. In
the meantime, until someone does something (he said passing the buck), I
find that when I want to know what the truth may be, or to get as close to
the truth as is possible, the only solution is to go back, get the original
paper used as a reference, and read it for myself. Which is an enormous time
consuming pain. But I believe that references are now so badly corrupted
that it is virtually impossible to trust them, or the papers based on them,
anymore. In effect this means that the entire medical research machine is
close to meltdown. If it hasn't melted down already. In
the meantime, remember that the truth is out there. It is just extremely
difficult to know what it is any more. More essays by Malcolm Kendrick ATKINS
AND THE FIRST LAW OF THERMODYNAMICS
‘The change in internal energy of a system is equal to the heat
added to a system minus the work done by the system.' Please
don't get me wrong. I am a great supporter of the Atkins diet. Anything that
helps to demolish the myth that eating animal fat, or saturated fat, causes
health problems gets my vote every time. And I do believe that many people
who try the Atkins diet do lose weight – when no other diet has worked. But
I want to tackle a myth about the Atkins diet which seems to have taken hold
of people's minds, causing them to believe in magic. Namely, that you can
eat as much protein and fat as you like and still lose weight. It is those
terrible carbohydrates that turn to fat and make you fat. Of
course if you eat fat, it doesn't need to turn to fat, because it already is
fat. And once you eat it, it is transported straight to the fat stores in
your body, where it is stored as….you got it… fat. Quite how fat
converted from carbohydrate is worse for you than fat that comes straight
from fat escapes me. Equally,
one gram of fat contains twice as much energy as energy as one gram of
carbohydrate. So if you eat more fat, by weight, than carbohydrate, you are
taking in more energy and should get fatter. But
somehow people seem to fervently believe that there are some processes going
on when you eat the Atkins diet that somehow allow the body to dispose of
excess energy gained from eating fat – in some way. However, if you eat
carbohydrates these will….what? Gather energy from another dimension and
convert this excess energy to fat? The
reality is that, when it comes to energy, the body is like any other system.
It loses energy by radiating energy to the surrounding environment, or doing
work. And the only way that the human body can radiate energy is as heat. We
don't produce light, electromagnetism, radioactivity, or very much in the
way of sound. For
very good reasons we don't excrete excess energy either. It is true that
some food passes straight through without being absorbed, but not much. If
you are diabetic, you can lose some sugar through the kidneys, and a few
ketones can escape here or there. But the body is a complete miser where
energy is concerned. What comes in stays in. And
the only way to increase energy output is to increase work. So
to suggest that you can eat as many calories as you like – in the form of
protein and fat – and still lose weight is quite frankly nuts. If the
Atkins diet can achieve this, then the single most important law of physics
is wrong. If energy can just disappear from the body, then it is
departing this Universe and going somewhere else. So, someone in a parallel
universe must be getting mightily pissed off that they are eating nothing,
and still putting on weight. (I know how they feel). I
like to think that I have an open mind. But even I draw the line somewhere,
and in this is case it is here. Energy cannot disappear. Full stop. Atkins
zealots have tried to explain this, the inexplicable, in a variety of
increasingly desperate ways. It is claimed that it is much more complicated
to turn fat into energy, so it takes more energy to do this than it does to
convert sugar into energy. It's
true, converting fat into energy takes several more steps. Which probably
does use up more energy. What's the point being made here? That burning fat
takes up more energy than burning up sugar, so….. The body uses more
energy burning fat. And what would happen to this excess energy? It would be
used to heat the body. Jolly good, that's mainly what we use energy for. Others
have said that on the Atkins diet you start to excrete ketones, and as
ketones are molecules that contain energy this is one way that the body
sheds excess energy on the diet. I have never believed this, but it is
possible I suppose. However,
I watched a programme on the Atkins diet a couple of nights ago. On this
programme they got identical twins and put one on the Akins diet, and the
other a low fat diet. They then stuck them both in sealed rooms where all
energy expenditure could be monitored. Oxygen use, loss of ketones in urine
and breath. They collected everything, which sounds a bit yucky, but there
you go. The
findings were that over a two week period the twin on the Atkins diet lost
approximately one extra calorie in ketone bodies. Equivalent to about one
grain of sugar. Which is exactly what I expected. The
reality about the Atkins diet is that, firstly, it works for many people.
Secondly, it does not cause any health problems, as far as I'm concerned.
Thirdly, it can only work because people cut back on their energy intake.
Because, to quote Scotty of the Starship Enterprise. ‘You cannae change
the laws of physics Captain.' Energy cannot just disappear. More essays by Malcolm Kendrick
Dec
3, 2003
STATINS
FOR CHILDREN – THIS IS MADNESS Here’s
the offending headline: ‘Despite
Controversy, Pressure Grows to Treat High Cholesterol in Children After
Studies Link Elevated Levels to Adult Heart Disease.’ The
first thing that I have to point out here is that, in primary prevention
trials, statins have never been found to reduce the risk of death. I don’t
care if they have been found to reduce the rate of heart disease. Does it
really matter if someone is saved from dying of heart disease, only to die
of something else? By
primary prevention trials, I mean trials in people who, whilst they may have
risk factors for heart disease, have not been found to have any clinical
signs, or symptoms related to heart disease. Secondary
prevention trials are different. These are done on people who have already
suffered a heart attack, or have angina, or some other clinical
manifestation of CHD. And it is true that in ‘secondary prevention’
trials, statins have been found to reduce the rate of dying of heart
attacks, and also to reduce overall death rates. By a small, but significant,
amount. However,
that is not relevant to this discussion. Because, by definition, all
children are in the primary prevention category. And this means that there
is not one scrap of evidence to suggest that statins will do them any good.
The best you might manage is to shift their cause of death from heart
disease to something else – usually cancer – about sixty years in the
future. How
do I know this? Because the clinical trials tell me so. If
we look at five major primary prevention trials: PROSPER, ALLHAT, WOSCOPS,
ASCOT and AFCAPS. (Don’t worry about the acronyms, they are not important,
they are just supposed to make the trials memorable). We can pull them apart
to look at the figures. By
the way, if you want to check my figures visit The Therapeutics Initiative
at The University of British Columbia http://www.ti.ubc.ca/ and look for Therapeutics newsletter number 48. Or, get the data from
the trials themselves. These
five trials had, between them, over forty thousand patients enrolled. Most
of them lasted at least five years, and they have all been endlessly quoted
in the medical literature. In short they are big, important and influential. So,
what was the overall mortality rate in those given statins versus the ‘control’
population? Morality
in those on statins was 6.6% And
what was the percentage of serious adverse events (SAEs)? A serious adverse
event is something like developing cancer, or having a non-fatal MI, or a
non-fatal stroke. So, pretty damned serious. In
fact, only two of trials reported this, as the majority of statins trials
keep quiet about SAEs. Serious
adverse events in the control population was 43.9% I
suppose you may be thinking, my goodness, there was a 0.3% reduction
in overall mortality. It may be small, but it’s still there. True. However,
although these five trials are usually presented as purely primary
prevention trials, they all included a secondary prevention population, 18%
on average. This more than accounts for any difference in overall mortality.
Even
if it doesn’t. I must point out that the difference is not large enough to
discount the possibility that this was merely a chance finding. These
figures do not get anywhere near statistical significance - the holy grail
of clinical trials. In
addition to this, the 0.3% reduction, if it really exists, took five years
to appear. Which means that, even if you take the best case scenario
possible, and ignore the fact that any difference is most likely due to
chance, you would have to a take a statin for fifty years to reduce your
risk of dying by 3%. At the same time, of course, you would have a 3%
greater risk of suffering a serious adverse event, such as a stroke, or
developing cancer. Does
this really represent powerful enough evidence to warrant starting a
four-year-old child on statins, and keeping them on for the rest of their
life? I
don’t think so. Especially not in the case of this Washington Post
reporter. For, in her article, she was using the example of a four-year-old
girl. And what do the statin trials tell us about the benefits of statins in
primary prevention in girls, or women? According to The Therapeutics
Initiative group: ‘There
were 10,990 women in the primary prevention trials (28% of the total). Only
coronary events were reported for women, but when these were pooled they
were not reduced by statin therapy. Thus the coronary benefit in primary
prevention trials appears to be limited to men.’ What
the statin trials tell us about women is that, in primary prevention,
statins can’t even manage to prevent heart disease, let alone anything
else! Has
the world gone completely mad? Are we really suggesting that we should start
a healthy four-year-old girl on a medicine, and continue this medicine for
the rest of her life? Something that could turn her into one of the ‘worried
well’, and even if it doesn’t, will most likely cause side-effects. Can
we really be contemplating this, when all of the evidence that exists points
to the fact that STATINS WILL DO HER ABSOLUTELY NO GOOD AT ALL! Apparently,
we are. ‘Anyone for tea?’ Asked the Mad Hatter. More essays by Malcolm Kendrick WHY
THE ATKINS DIET IS HEALTHY A
man called Banting promoted a diet pretty much indistinguishable from that
of Atkins in 1863. In fact, the verb to ‘bant' is used in Sweden as a term
for going on a diet To
find out more about the Banting diet (now known as the Atkins diet) go
here Anyway,
reasonably balanced or not, on this programme there was still an
unquestioned view that, even if the Atkins diet did help with weight loss,
it was still damaging to health. It would cause kidney disease, and
osteoporosis and heart disease. Various professors of nutrition were wheeled
out to condemn the Atkins diet as dangerous nonsense. Ignoring
the kidney disease and the osteoporosis for now, the nutritional professors
made the usual statements. For example, ‘It is known that saturated fat
increases the level of blood cholesterol and causes CHD.' They didn't quote
any evidence for this. As far as they were concerned it is just a known fact. Well,
what is the evidence that a diet high in saturated fat raises your
cholesterol level? Where does it come from? The Framingham Study? That world
famous study that is quoted by medical experts around the world. "In
Framingham, Massachusetts, the more saturated fat one ate, the more
cholesterol one ate, the more calories one ate, the lower people's serum
cholesterol...” Dr William Castelli 1992 (Director of the
Framingham study) So
the evidence obviously didn't come from Framingham. What about studies in
children? These poor vulnerable imps, where the damage is first being
done? Just to get a bit of genetic diversity into the equation, let's look
at Chinese children first. ‘Children
in the intervention group were fed with low-cholesterol and low-saturated
fatty acid diet, and the control group with normal diet. The duration of
intervention was three months. Compared with the control group, serum
cholesterol levels of children under intervention were not significantly
changed. Total cholesterol: 4.64 (186dg/ml) vs 4.68 (188dg/ml) mmol/L LDL:
2.66 (107dg/ml) vs 2.62 (106dg/ml).' Zhu WL et al Zhonghua Liu Xing Bing
Xue Za Zhi. 2003 Sep Then
children in the UK: ‘Unexpectedly,
significant inverse associations were found between the dietary content of
saturated fatty acids on the one hand and the serum concentrations of
cholesterol… on the other.' Samuelson
G et al Br J Nutr Mar 2001 The
reality is that, in many different studies, it has been shown that the more
saturated fat you eat, the lower your cholesterol - although the difference
is not that great. Of potentially greater importance is that a high fat diet
has a more significant effect on raising HDL and lowering VLDL. Which is
supposed to be very healthy indeed. Consider
this extract from the University of Pennsylvania: I'm
sorry that I can't present you with anything much from PubMed (the bible of
mainstream medical research) about this. But as others may have discovered,
any paper that supports the Atkins diet has no abstract attached in PubMed
– you just get blanks. Did someone use the word censorship? Not me your
honour. I would never dream of saying such a thing. Now,
anyone who has read my scribbles before will realise that I don't think the
level of any lipid in your blood makes the slightest difference to the rate
of CHD. But most other people do, so I think it is worth explaining why a
high fat diet will automatically raise HDL and lower triglycerides. A
fact, by the way, that seems to have created stunned surprise amongst many
researchers when results from the Atkins diet were published. Which just
shows that they need to go back and read their textbooks again. In
order to understand why a high fat diet should, and does, raise HDL levels
and lower VLDL levels (and may also lower LDL levels), you need to
understand a bit about fat and sugar metabolism and the role of lipoproteins
in your blood. Starting here. When
you eat fat it is absorbed by the gut and stuffed into very large
lipoprotein known as a chylomicron. The fat in a chylomicron is almost all
stored in the form of three fat molecules attached to a glycerol molecule, a
structure known as a triglyceride. Three fats and a glycerol = tri-glyceride.
By the way, cholesterol also sits in chylomicrons as a co-passenger. (Anything
insoluble in water/blood, such as cholesterol, has to be carried around in a
lipoprotein) Chylomicrons
are then released into the bloodstream and travel through the body losing
chunks of triglyceride all the while as they pass fat cells. (Fat cells
attack chylomicrons with a ‘lipase' enzyme, chopping bits off). As this
happens chylomicrons shrink, turning into Very Low Density Lipoproteins (VLDLs),
which are otherwise known as… ‘triglycerides.' How confusing is that? In
fact, the nomenclature in this area must be the most confusing in all of
medicine.
It's
little wonder that most people haven't the faintest idea what anyone is
talking about in lipid metabolism. Chylomicrons, VLDL, HDL and LDL are all
lipoproteins. I wish that people would stop calling them things like ‘cholesterol'
and ‘triglycerides', and ‘good' cholesterol and ‘bad' cholesterol. It
really doesn't aid understanding. Anyway,
moving on. Apart from chylomicrons, the gut also sends out VLDLs de-novo,
and the VLDLs do pretty much the same thing as chylomicrons, dropping off
triglycerides here and there (mainly into fat cells) and shrinking. Quite
what the difference is between a shrunk down chylomicron and a VLDL is, I
don't know. (By the way, just in case you're wondering, VLDLs also contain
cholesterol as a co-passenger. All lipoproteins have cholesterol in them) Not
all chylomicrons and VLDLs travel round dropping off triglycerides. Some go
straight to the liver where they are absorbed, broken down, and unpacked.
And their contents are used to make other things the body needs. However,
wherever they go, all of the ‘fat containing' chylomicrons and VLDLs
produced by the gut drop off their fat load, shrink, are then absorbed and
completely disappear. So a few hours after a meal they are gone. And if you
were to measure VLDL levels a few hours after a high fat meal they would
have returned to ‘normal'. Whatever normal may be. Thus,
if you eat a high fat meal, almost all sign of it will have disappeared in a
relatively short space of time. And there will be no change in any lipid
level. Or at least not any lipid level that anyone can be bothered measuring. However,
if you eat a high carbohydrate meal, the metabolism acts in a very different
way. Carbohydrates are absorbed and transformed into sugars in the gut, from
whence they go straight into the bloodstream, same as fat. But because
sugars are soluble in water they don't need to be carried in a lipoprotein,
so there is no immediate effect on lipid levels from a high carb meal. You
just get a sharp rise in blood sugar level. A
certain amount of the sugar will be absorbed into fat and muscle cells, and
then stored as glycogen. But if you eat a big carbohydrate meal, the fat and
muscle storage cannot cope, and the excess sugar has to be absorbed by the
liver to prevent the sugar level getting too high. However,
the liver cannot store that much sugar, so it starts to convert it into
fats, in the form of triglyceride. At which point, the liver then packs this
excess triglyceride into a VLDL and sends it out into the bloodstream -
along with some cholesterol. (Unlike with sharks, the liver in humans is not
an energy storage organ) So
you get a kind of delayed VLDL rise after eating carbohydrates. But there is
a key difference between the VLDL made by the guts, and the VLDL made by the
Liver. The VLDL made by the liver, unlike that made in the gut, shrinks into
a low density lipoprotein (LDL). The dreaded heart disease causing
lipoprotein – the one they call co-lest-erol. Why
does this happen to ‘liver manufactured VLDL', when it doesn't happen to
the VLDL made in the gut? Well, as liver manufactured VLDL leaves the liver,
it interacts with an HDL molecule which transfers it's proteins to the VLDL
molecule. One of the proteins transferred is apolipoprotein B-100. And the
apo B-100 molecule is the unique LDL ‘identifier.' On
the other hand, VLDL made in the gut has apolipoprotein B-48 attached to it
and this VLDL doesn't become an LDL molecule as it shrinks. Now,
if you are not already completely confused, I will explain what this means. Rewind.
If you eat fat, it is absorbed from the gut, packed into chylomicrons and
‘VLDL B-48s,' and transported around the body and then got rid of. Gone.
So immediately after a high fat meal you will have a very high triglyceride
level, made up of VLDL B-48, but this will fall relatively rapidly.
Importantly, there can, and will be no effect on HDL or LDL levels. And so
if you measure the lipid levels in the fasting state (which is when such
things are measured) you will find nothing at all after a high fat meal. On
the other hand, if you eat a high carbohydrate meal, the level of VLDL B-48
will not rise. But some time later, the liver will start converting excess
sugar into fat and sending this out in VLDL B-100 molecules. And this
process can go on for many hours after a meal. So the VLDL level may still
be high when you measure it. In
addition to finding a high VLDL you should also find a low HDL. Because, for
each VLDL the liver makes, an HDL hands over its proteins and disappears. So
the more VLDL the liver makes, the less HDL you will have. Cause and effect. Also,
as you may have noted. If the VLDL B-100 all ends up as LDL, the more VLDL
the liver makes, the higher the LDL level is likely to be. Therefore,
if someone is on a high carbohydrate diet, they should automatically have a
raised VLDL level, a reduced HDL level and quite possibly a raised LDL level. Golly
gee whiz. A high fat diet reduces VLDL, raises HDL and may even lower LDL.
And a high carbohydrate diet does the exact opposite. In short, the
metabolism does exactly what you would expect it to. So
you see. Atkins was right all along. Even if he didn't appear to know why. More essays by Malcolm Kendrick LIQUID
DRANO-LIKE SYNTHETIC “GOOD CHOLESTEROL” PLAQUE BUSTER! IT’S SCIENCE,
BUT NOT AS WE KNOW IT
‘A
synthetic form of "good" cholesterol has been shown to quickly
shrink blockages clogging coronary arteries, offering for the first time the
possibility of a drug that could actually rapidly reverse heart disease,
researchers reported yesterday….' I'm
writing a book at the moment called Cholesterolmania. That plus a job, plus
children and home, an attempt at a social life and columns at
redflagsdaily.com. That's a tad busy, and I thought I'd take a short break
from column writing, but…I couldn't let the above story from the Washington
Post go without comment. Here
is my immediate response. Aaaaaarrrrrrggggghhhhh! Thud. It's
almost impossible to know where to start without ranting. Firstly, just to
clear something up, HDL is not cholesterol ‘good' or otherwise. HDL,
stands for High Density Lipoprotein. It is a lipoprotein that is
manufactured in the guts and the liver, and it contains a small
amount of cholesterol. HDL
appears to have two basic functions in the body. Firstly, it transfers
proteins, known as apolipoproteins, to VLDL, allowing the VLDL to be
recognised by receptors around the body. Secondly, it removes cholesterol
that is floating about and takes it back to the liver. When
cells do break down in body, which is happening all the time, the
cholesterol from cell walls is released into the surrounding extra cellular
fluid. The HDL lurking in the vicinity ‘mops up' this excess cholesterol
and transfers it back to the liver. This scavanged cholesterol is then used
in the manufacture of Very Low Density Lipoproteins VLDLs. VLDLs contain two
basic ingredients, fats (in the form of triglyderides) and cholesterol. VLDLs
are then sent back out into the bloodstream. As VLDLs lose triglycerides
they shrink in size, becoming Low Density Lipoproteins LDLs (otherwise known
as ‘bad' cholesterol – for some stupid reason). LDLs are then absorbed
by cells that need cholesterol, and the cholesterol is unpacked and used to
build various structures within the cell, including the cell wall. Which
means that HDLs are part of a re-cycling mechanism for cholesterol. At the
risk of repeating myself, the liver manufactures cholesterol and sends it
out within VLDLs. As VLDLs lose triglyceride – which provides energy for
cells around the body - they shrink into LDLs, and LDLs are then absorbed
into cells where the cholesterol is unpacked. When
a cell then dies, it releases cholesterol, which is mopped up by HDL and
transferred back to the liver. This is not immensely complex, but for some
reason, mainstream researchers have decided that HDL can, in some way,
protect against the build up of atherosclerosis. There
are two reasons for this, I think. Firstly, because a low HDL level seems to
be an important risk factor for CHD, even more so than a raised LDL level
(So surely it must be doing something…Duh!) Secondly, because it has been
noted that, as HDL does indeed transfer cholesterol from around cells and
back to the liver, it is thought that this reverse cholesterol transport
might, in some way, be able to suck cholesterol out of atherosclerotic
plaques. In
answer to the first piece of stupidity. If VLDL levels go up HDL
automatically goes down, it's all to do with the transfer of apolipoproteins
from HDL to VLDL. The raised VLDL itself is caused by underlying insulin
resistance – one of the basic causes of heart disease. So a low HDL is
merely a ‘marker' for raised VLDL, which itself is a marker for insulin
resistance. A low HDL by itself causes nothing and prevents nothing. With
regard to the reverse cholesterol transport nonsense. HDL cannot, I repeat
cannot, remove cholesterol from atherosclerotic plaques. It is impossible
for this to happen. The cholesterol in a plaque is not ‘floating free' in
the extra cellular fluid. It is trapped in a solid atherosclerotic lump. HDL
is completely and utterly incapable of getting at it, and even if it could,
it could not separate it out from the surrounding plaque structure. HDL is a
passive inanimate chemical. It cannot carry out complex tasks. The
concept that HDL could remove cholesterol from a plaque is such a stupid
idea that I cannot believe it still exists. Once you understand the science,
the whole thing is patently ridiculous. If
synthetic HDL can reduce the size of plaques then I will eat my hat. What
these researchers are seeing, probably, is what all researchers see. Most
plaques, if left alone, do gradually reduce in size – a bit. Alternatively,
they have been looking at their findings with eyes of faith. Let's just see
if anyone else can verify these results. P.S.
In the Heart Protection Study (HPS), a major study in which the rate of
deaths was reduced in patients taking a statin (simvastatin), at post-mortem,
the people who had been taking the statin had bigger and more complex
plaques than those who had not. In reality, the size of the plaque does not
actually have anything to do with how dangerous it is. More essays by Malcolm Kendrick TELEOANALYSIS — OR WHEN I FINALLY REALIZED THAT I HAD FALLEN DOWN THE RABBIT HOLE
After
reading a paper in the British Medical Journal which appeared a few days ago,
I didn’t know whether to laugh or cry, or stand at the edge of a cliff and
scream. Instead I thought I would write a column, so that you may share my
sense that the world has finally gone completely bonkers. The
paper was called: ‘Teleoanalysis
— combining data from different types of study’ Which
sounds pretty unremarkable, and contains seemingly sensible remarks, such
as: ‘Teleoanalysis can be defined as the synthesis of different
categories of evidence to obtain a quantitative general summary
of (a) the relation between a cause of a disease and the risk of
the disease and (b) the extent to which the disease can be
prevented.’ My,
how reasonable this seems. Yes, of course, carry on — carry on. This is
mathematics isn’t it, or something of the sort? So, what comes next? ‘It
may also be necessary to quantify the individual effects that
relate to separate steps in a causal pathway–that is, the
effect of factor A on disease C is determined from the estimate
of the effect of A on an intermediate factor B and the estimate
of the effect of B on C, rather than by directly measuring the
effect of A on C. The exercise is like putting together the
pieces in a jigsaw puzzle.’ I
see, so A causes B, and B causes C. So it can be deduced that A causes C.
Bravo…. Well done. How simply splendid. Therefore,
we can use the following reasoning. A
high saturated fat intake (A) causes an increase in cholesterol levels (B)
(A causes B), a raised cholesterol level (B) causes heart disease (C) (B
causes C). Ergo, we know that a high saturated fat intake causes heart
disease (A causes C). You
may not think that there is anything much wrong with this. It sounds utterly
logical — doesn’t it. So, why is anyone bothering to write this article? Well,
you see there is a problem with the ‘saturated fat caused heart disease’
hypothesis. Namely, that no interventional trial has ever shown that reduced
saturated fat intake has any impact whatsoever on heart disease rates. (An
interventional trial is one where you ‘intervene’ and change something,
such as dietary fat intake — these are normally considered ‘gold
standard’ clinical trials). As admitted by the authors: ‘A
meta-analysis of randomised trials suggested that a low dietary
fat intake had little effect on the risk of ischaemic heart
disease.’ So
we have a problem. We ‘know’ that saturated fat raises cholesterol
levels, and we ‘know’ that raised cholesterol levels cause CHD. We just
can’t seem to show that if you lower the saturated fat intake you have a
reduction in the rate of CHD. Which would kind of suggest to most people
that A doesn’t cause B, and B doesn’t cause C. But
no, this cannot be true, this is wrong! Therefore, any results contradicting
this must be wrong. So there! It’s always reassuring when a scientist just
‘knows’ that something is true. It avoids all those tedious clinical
trials that are sometimes needed for proof. Anyway,
in order to prove that the interventional trials are wrong we use
teleoanalysis. The first step in teleoanalysis, apparently, is to condemn
all the trials that fail to show what you want, using statements such as:
‘But the effect of a significant reduction in dietary fat
can easily be underestimated, even when it is based on the
results of randomised trials.’ Then
we use the second step in teleoanalysis, which is that we to look at the
studies we want (A causes B, and B causes C — carefully ignoring all
studies that showed the complete opposite), and from that extrapolate the
answer to studies that have not been done, but had they been done,
would have shown exactly what we already know to be true. You think I am
joking? ‘…teleoanalysis
provides the answer to questions that would be obtained from
studies that have not been done and often, for ethical and
financial reasons, could never be done.’ It
is so much better, I find, to rely on answers from studies THAT HAVE NOT
BEEN DONE, and COULD NEVER BE DONE. In this way you can always get the
results that you want, and you never ever need to carry out any more studies
that might contradict the things you already know to be true because for
ethical and financial reasons these trials never can be done. Truly…
I mean. Gasp….thud. When I read this, I thought it must be a joke. But
this was written by one of the authors of the infamous Polypill article,
suggesting a one-trick multiple pill could prevent heart disease. The man
who also wrote the ‘time-lag hypothesis,’ explaining that the French don’t
have much CHD because they haven’t been eating as much saturated fat as
people in the UK and USA, at least not before 1970. Are
there any limits to the double-speak that can be used to prop up the
diet-heart hypothesis? Apparently not. Perhaps I will wake up and find this
is all a dream, for right now I do feel as if I have fallen down the rabbit
hole. P.S. If you think I have made this up as a joke, I refer you to the BMJ article.
ATKINS MUST BE DESTROYED!
I
am a great fan of the science philosopher Karl Popper, that is whenever I
can manage to understand what it is that he is saying. I get through one
paragraph at a time, very slowly, then I have to go and lie down until my
brain stops hurting. Popper
has much to say on the theme of science, scientific progress and the like.
He was highly pro-science and the proper use of the scientific method. But
he was also acutely aware of the danger that science, and scientists, could
become so entranced by a hypothesis that it became the answer, the
truth, a belief. And those who dared to question such a fundamental belief
were metaphorically burned at the stake. In
fact, if you were to share my interest in the development of scientific
thought, it is clear that the big breakthroughs, the things that we all now
accept as true e.g. the theory of evolution, were generally forced through
against the might of the prevailing scientific orthodoxy. And, in general,
the famous scientists that you have heard of e.g. Darwin, usually followed a
few others who were crushed so effectively that there names have vanished
from the record. Darwin
was far from the first to propose the theory of evolution. However those,
like Chambers, who promoted the idea before Darwin were cut down and
humiliated by the leading scientists of the time who believed that a species
could not change into another species — for some unfathomable reason or
another. Darwin bided his time, and let a few others line his path to glory
with their broken reputations. This
happened about one hundred and fifty years ago. However, with regard to
unquestioned dogma, things are much the same today. As I write this, there
are new hypotheses that cannot be questioned. For example, the hypothesis
that mankind is causing global warming by burning fossil fuels. Or, that we
are creating a massive hole in the Ozone layer by use of CFCs. The hole in
the Ozone layer closed up last year by the way — but you probably never
heard much about that. Anyway,
dare to question those two orthodoxies and you will receive hysterical abuse.
Everybody just knows that the world is warming up, and we are ripping a
great hole in the Ozone layer. This may or may not be true, but neither
belief is based on rational thought. They are driven by rather deeper,
emotional beliefs. As an aside, when you find yourself really wanting to
believe that something is true — then it usually isn’t. However,
over to Atkins. Current scientific orthodoxy has decreed that a high fat
diet, especially saturated fat, is bad for health. It raises cholesterol,
kills us from heart disease, and causes breast cancer and all sorts of other
nasty things. For many, and especially those at the heart of the medical
community, this is a Truth that cannot be questioned. And a huge scientific
and financial structure has grown up around this Truth. But
in the last few years Dr Atkins and his diet have begun to make significant
inroads. Now, I know that people generally use the Atkins diet for weight
loss, and not any other health promoting reasons. But the Atkins diet was
virtually heresy. Here was a man saying that if you ate saturated fat you
would lose weight and be healthier. People taking the Atkins diet even had
the cheek to find that their cholesterol levels dropped. Shock horror. At
first the scientific community used the first of the immunizing tactics that
scientists use to defend sacred beliefs, as defined by Popper: Immunizing
tactics 1:
Ignore the refutation So
at first Atkins was ignored, but he would not go away. The mainstream
medical church then said that the Atkins diet was not actually high in fat,
or at least not the really damaging sort of fat. But it was, so that didn’t
work. They then said that there was a huge bulk of evidence to support the
hypothesis that saturated fat raised cholesterol levels, and that Atkins was
wrong. But he wasn’t, and the trials set up to show that the Atkins diet
was dangerous were hopeless failures, in that these trials confirmed what
Akins had said all along. People on the Atkins diet achieved a reduction in
cholesterol levels (not that I believe this matters a tin of beans). Where
next to support the diet/heart hypothesis. The ‘hypothesis that is the
truth,’ as revealed to us by Ancel Keys. Where next indeed? After failing
to show terrible dangerous levels of raised blood cholesterol, or any other
nasty things, came the personal attacks. Atkins is a dangerous man promoting
a highly dangerous diet. Atkins is not a scientist, how can he know anything.
Aktins cannot prove the benefits of his diet, he hasn’t done the required
research (mostly true, by the way). Next
came the terror tactics: the Atkins diet is dangerous and damages your
kidneys; makes your blood acidic; it upsets your metabolism; and, in fact,
people are dying on the diet. This
is all complete rubbish. Whilst it is true that a diet high in fat and
protein will result in greater production of acidic residues, ketone bodies
and the like, and your blood and urine will become slightly more acidic,
there is not the slightest, remotest, teeny weeniest piece of evidence to
support the claim that this is in any way damaging. Not a single clinical
end-point has ever been demonstrated to be affected. And
if you read anything that does claim the Atkins diet is damaging, please
take the time to read the small print — if you can find it. What you will
normally discover is that the level of some substance in the blood is found
to be raised which may (note the word may), lead to kidney damage.
But by the time this reaches the newspapers it has been converted into ‘Atkins
diet kills thirteen year old girl by destroying her kidneys.’ Perhaps
those leading the Atkins attacks would care to raise their gaze up to the
Innuit in Canada and the frozen north. In years gone by they rarely ate a
vegetable, any fruit or a carbohydrate molecule. They existed almost
entirely on fat and protein. When they were studied (before their lifestyle
changed), they were found to be in exceptional health. Without, it must be
added, any sign of heart disease or renal failure. But
my point, the point of this column, is not to discuss whether or not the
Atkins diet works. I wanted to make it clear that the attacks on Atkins are
not scientific, not rational. Atkins, may he rest in peace, is being
attacked because his diet threatens the mainstream. He, and his supporters,
are being subjected to the secular equivalent of the Spanish Inquisition.
‘Admit that you are wrong, or I shall destroy your reputation.’ None of
the attacks on Atkins are scientific, none of them are correct. They are an
attempt by the ‘herd’ to protect the sacred diet/heart hypothesis. The
sort of attacks that Popper would have recognised for what they are. Atkins
must be destroyed to protect the mighty diet-heart hypothesis. And all of
the recent articles that just seem to be springing out of nowhere about the
terrible dangers of the Atkins diet are designed to do just this. More essays by Malcolm Kendrick SO,
WHAT DOES CAUSE HEART DISEASE? When
you have spent twenty years of your life studying something, you can become
somewhat of a bore on the subject. But please bear with me, because I am
going to reveal to you the true cause of Coronary Heart Disease (CHD). A
bold claim indeed, but I think I can sustain it. The
first thing to state, however, is that there is no single cause, no one
factor. If there was, it would have been discovered by now. I sometimes
think that the obsession with finding the cause of a disease has
seriously hampered research into this, and many other areas. There is always
a sense, within science, that the answer, when you find it, should be
simple, and that therefore the simplest explanation is usually correct: E =
MC2 and all that. Occam’s razor, terribly seductive, but not
always true. Another
thing that has seriously hampered heart disease research is the factor that
I call "terminological inexactitude." In CHD research, the words
heart disease, CHD, CAD, CV disease are all whirled about almost
interchangeably. Equally, various papers talk about atherosclerosis. But the
term doesn’t actually mean anything. If
you describe atherosclerosis as thickening and hardening of the arteries,
then almost all populations throughout the world suffer from the same rate
of atherosclerosis. Yet, the rate of CHD between populations can vary more
than fifteen-fold. Which means that atherosclerosis isn’t the underlying
cause of CHD, at least not if you define atherosclerosis as anything that
isn’t a perfect, springy, smooth artery. In
order to understand CHD (another horribly inexact term), you must be a bit
more precise about what it is that you are actually talking about. And what
I am talking about are discrete, or focal, areas of arterial damage. Some
people refer to them as plaques, and so that is the term I will use. Plaques
are the little beauties that can narrow an artery, causing things like
angina. They can also ‘rupture’ causing a blood clot, and then complete
obstruction to blood flow within an artery, leading to myocardial infarction,
or a stroke (or problems elsewhere). You can have as much thickening, or
hardening, or atherosclerosis of the arteries as you like. But if you haven’t
got a plaque or two, you will not die of CDH. So,
what causes plaques to develop? There
are two basic processes that do this
The
two processes are highly interconnected. For example, damage to the
endothelium stops it from acting as an anti-coagulant surface, making it
more likely for a thrombus to form over the damaged area. When
a blood clot, or thrombus, forms over an area of artery wall, this is the
start of plaque formation. Repeated thrombus formation over the same spot
causes the plaque to grow, and eventually it can completely block the artery. Therefore,
any ‘factor’ that causes either endothelial damage or increased blood
coagulability will increase the risk of dying of CHD. Factors
that have been shown to damage the endothelium include:
Factors
that make the blood more prone to clotting include
These
lists are not exclusive, but they highlight the main factors. You will have
noticed a great deal of overlap, which is not surprising. Why
some things protect against heart disease If
you look at those lists, it becomes clear that things that protect the
endothelium and/or reduce blood clotting should reduce the risk of heart
disease. So, what things do protect against CHD, and how.
Equally,
what factors cause plaques to develop? Some
of them are the usual suspects:
Whether
or not a raised blood pressure can cause plaques to form is a moot point.
One can see that a high blood pressure may cause endothelial damage, but the
evidence from blood pressure lowering trials shows zero impact on the rate
of death from CHD So, I think the jury is out on this one. There
are other factors that are relatively rare that directly cause CHD. Such as:
The
single most important cause of CHD, however, is metabolic syndrome. This
syndrome can be caused by a number of different factors:
Whatever
the cause, metabolic syndrome develops because of abnormal cortisol levels.
The abnormal cortisol levels, in turn, cause insulin resistance (cortisol is
a powerful insulin antagonist). This then leads to a spectrum of metabolic
abnormalities:
As
previously described, these factors then lead to plaque formation. It
is likely that the most common cause of metabolic syndrome is chronic
stress, which creates HPA-axis abnormalities (the HPA-axis is the system
that controls the reaction to stress) and then abnormal cortisol secretion.
Populations most likely to suffer from chronic stress are those suffering
social dislocation, emigration, forced relocation, etc. For
this reason migrants will generally have high rates of CHD. Also populations
who have been disrupted by other populations moving in on top of them, e.g.
Australian aboriginals, Native Americans, Maoris in New Zealand, etc. In
addition, populations undergoing rapid social change will suffer from CHD.
This is currently true of Eastern Europe where the rate of CHD has exploded
since the breakdown of the Soviet Union. In
support of this general hypothesis, the population group with the highest
levels of heart disease, currently, are ‘Asian Immigrants,’ i.e. those
who have emigrated from countries such as India, Bangladesh, Pakistan.
Wherever Asian immigrants move to, they suffer very high rates of heart
disease. Additionally, they have a very high rate of metabolic syndrome, and
high levels of cortisol secretion. And just to treat you to one quote on
this: ‘The
cardiovascular risk factors which comprise the metabolic syndrome are
associated with increased hypothalamic adrenal axis (HPAA) activity in some
Caucasian populations. South Asians have high rates of cardiovascular
disease and its risk factors. We investigated the relationships between HPAA
activity, adiposity and the metabolic syndrome….. ….This
study demonstrated that fasting 09.00h cortisol concentration is strongly
associated with cardiovascular risk factors in a South Asian population.’ Ward
A.W et al: Clinical Endocrinology (2003) 58,500-505. Another
important cause of heart disease is eating food under stress. If you eat
whilst under physical or mental stress, you will be producing stress
hormones: cortisol, adrenaline (epinephrine), glucagon and growth hormone.
These are all powerful insulin antagonists. The
antagonism from these hormones during a meal will lead to spikes of blood
sugar, insulin and triglyceride (to name but three factors) as insulin, the
anabolic hormone, battles against the catabolic stress hormones. Which
is why a country such as France — which has the same level of ‘classical’
risk factors as the USA — has a low rate of death from heart disease. The
French spend a long time eating their meals, so they give their metabolism a
chance to absorb and digest food properly, rather than set up a metabolic
battleground with stress hormones. So,
there you have it, now you know what causes heart disease. And if you want
to protect yourself against heart disease, do the following things:
P.S. What about high cholesterol levels? Well, what about high cholesterol levels? This red-herring has thrown researchers off the scent for the last sixty years. Only when it is abandoned as a risk factor will mainstream researchers be able to make sense of heart disease. More essays by Malcolm Kendrick
HOW
RISKY IS A RISK? I
have only just recovered from the idea that everyone in the whole world over
the age of fifty-five should spend the rest of their lives on six different
medications, all stuck together in one great big pill. You may have seen the
non-story about the non-existent polypill peddled in the British Medical
Journal (BMJ). I was stimulated to look again at the concept of risk. The
authors of the madcap polypill article in the BMJ made the claim that taking
their polypill would reduce the risk of dying of coronary heart disease (CHD)
by 80%. Whether or not you believe their figures — and I don’t — I
sense that this figure of 80% would be taken by most people to mean that
eighty out of one hundred people would be saved from death. Yet,
that is not what it means at all, for this figure is a relative risk
reduction figure. And a relative risk reduction means nothing, by itself.
However, because everyone’s eyes glaze over whenever you start talking
about statistics, most researchers manage to get away with using relative
risk reduction figures when, in reality, they should be shot for doing so. Now,
here’s a challenge. The challenge to make an article about statistics
interesting….. Okay, that’s not possible. But maybe a little bit
interesting? When
you talk about a risk, you need to know the absolute risk of a thing
happening. For example, the risk of getting struck by lightening. I don’t
actually know what that risk is, but I would imagine it is about one in five
million. But again, that figure means nothing unless you put a time scale on
it. Is this a one in five million risk over a hundred years, or one year? If
you don’t put a time scale in, you can claim anything you like. For
example ‘The Earth will be hit by a big Asteroid. This is one hundred
percent certain — shock claim from Astronomer.’ Read all about it. And
of course, this is true. The Earth will be hit by a big Asteroid, sometime
in the next three billion years or so. The odds ratio for this event is 1 =
100% certain. I am even willing to take a bet on it. So,
I must define risk in two ways, the possibility of the thing happening, and
the time period during which that thing will happen. With lightening
strikes, this is about a one in five million risk, over a five year period.
Not high. However,
people don’t tend to bend statistics by ignoring the time factor that
often. Unless they want money to fund their Asteroid defence system. A snip
at five trillion dollars, plus VAT. What generally happens is that people
inflate the risk in the following way. For example, the chances of dying of
lung cancer, for a non-smoker, are about 0.1%, lifetime risk. If, however,
you live with a heavy smoker, your chances will increase to about 0.15%. (These
figures are for illustration only). Now
you can report this in two ways. You can state that passive smoking can
increase the risk of lung cancer by 0.05% - one in two thousand. Or, you can
state that passive smoking increases the risk of lung cancer by fifty per
cent. Both figures are correct. If you are an anti-smoking zealot, then I
would imagine you would prefer to highlight the second figure. The relative
risk figure. And
when it comes to reducing cardiovascular risk, exactly the same procedure is
used (in reverse). Let’s say that the chance of dying of CHD over five
years, in a healthy fifty-five year-old, is 1%. By reducing this risk to
0.2%, you will have reduced the relative risk of dying of CHD by 80%. In
this way a 0.8% absolute risk reduction is hyped up as an 80% reduced risk
of dying of CHD. Mangling statistics is easy when you know how. It’s even
fun. Anyway, now you know the difference between a relative risk and an absolute risk, and I hope this makes it easier for you to hack your way through the misinformation that spews forth from the great medical research machine. BEWARE
THE PLACEBO EFFECT
Not that long ago, a number of people were put under general anaesthetic and
had holes drilled into their skulls. These procedures carried all the risks
of major surgery, yet the doctors who carried out the operations knew that
they would provide no benefit. Surely, you might think, this flies in the
face of the primary imperative of the medical profession ‘First, do no
harm.’ Why
was this done? It was done to satisfy the demands of the great placebo God.
A strange creature that lives in a metaphysical world. A creature of belief
without substance, a wraith like thing that constantly changes its shape and
appears in a different guise to everyone who sees it. But
surely, everyone knows that there is a placebo effect; it really exists.
Give someone a white pill with no active ingredients and it will have an
impact of some sort — usually a positive impact. Everyone believes this to
be true. So strong is this belief that almost all major clinical trials,
wherever possible, are split in two, with one group being given an active
substance, and the other group a placebo. If
you didn’t do this, you would not know if any improvement that you saw
from the drug was, in fact, due to the ‘placebo’ effect. So we are told. Ah,
the placebo effect. It is funny how, in science, there is normally some
attempt to define the impact of an ‘effect’ (e.g. the gravitational
effect). It would be difficult to imagine NASA sending up the space shuttle
without having made some effort to establish the escape velocity required to
break free of the Earth’s gravitational attraction. Yet,
everyone is quite happy to accept that there is a placebo effect, without
ever bothering to measure it in any meaningful way. It just happens, it’s
there, we all believe in it. We’ve all seen it, for goodness sake! Or have
we? Well,
let’s assume that I gave you a white sugar pill and said. ‘I am giving
you this pill, it is completely inactive, but you are supposed to think it
is going to do you some good. Do you understand… it won’t work, it is
completely ineffective.’ Do
you think that this would make the placebo effect more or less powerful? Has
anyone ever bothered to measure this… ah, well…. No. Never. Never ever. Most
people assume that the placebo only works if the patient thinks they are
taking a real drug. Has this ever been tested, or measured? Ah, well… no.
And, of course, the placebo has no ‘effect.’ A sugar pill cannot do
anything. It is only the belief in the pill that works. Or is it? Do we know? And
if there is a real placebo effect, should we not use it to treat diseases?
Actually, we can’t. Why not? Because the placebo effect doesn’t really
exist. Or, at least, it only exists if the patient really believes that they
are taking a real drug. Or does it? Most mainstream doctors believe that
alternative medicine works purely through the placebo effect. Is this true…
who knows? But
if it were true, what’s wrong with that. Just don’t look down, otherwise
you will be like one of those cartoon characters, suspended in air, until
they look below them and realise that there is nothing holding them up. At
which point…..aaaaaahhhhh! All
that we really know about the placebo effect is that, in some areas, such as
pain relief, patients who take a placebo report reduced pain, an effect as
powerful in some people as that achieved by strong painkillers. We have no
idea if there is any placebo effect in many other ‘objectively’
measurable areas, such as in cancer regression, wound healing, and
progression of coronary artery disease. Equally,
no study has ever been done comparing active drug, placebo and ‘doing
nothing at all’, to find out if doing nothing at all is equivalent to
placebo. No-one, in fact, really knows if a placebo arm is ever required in
any clinical trial. It is just an article of faith. And
to look at this another way: If clinical trials are supposed to reflect what
may happen in real clinical practice, no-one is ever prescribed a placebo in
clinical practice, they either get the drug, or they don’t. So surely, it
is important to establish the difference between taking a drug, and taking
nothing — as this will reveal the absolute benefit of taking the drug in
real life — even if that did include some placebo effect. So the placebo
arm offers no value, other than some data of purely academic interest. Yet,
to return to the start of this article, I was reading a recent trial on a
treatment for Parkinson’s disease which required implanting a major
electrical device in the patient’s brain. The placebo arm was required to
have surgery with holes drilled in their skulls, so that they could not know
that they did not have the device implanted….. Has
anyone ever established the maximum possible placebo effect in the treatment
of Parkinson’s disease? I think you may be able to guess at the answer to
this by now. In fact, some studies have clearly shown that there are
conditions with no placebo effect at all (whatever a placebo effect might
actually be), e.g. impact on major stroke. If Parkinson’s disease were one
of them, then a whole lot of people had holes drilled in their skulls for no
reason whatsoever. All
this, in reality, because a placebo can reduce pain. Big deal. It is hardly
surprising that the perception of pain can be altered enormously by telling
someone that ‘this white pill’ will reduce pain. I have never taken
morphine, or heroin. But I have spoken to those who have (for pain relief).
They have stated that they still knew the pain was there, but that it
didn’t bother them. Soldiers shot in combat often say that they feel no
pain at all. If
you cut off someone’s leg, they can suffer huge phantom pain from the leg,
with no leg present. The brain can create pain out of nothing, and also shut
it off completely. So
it is not exactly surprising that an inactive white pill presented as a
painkiller can reduce the perception of pain, or the recollection of it, or
the reporting of the recollection of it. However, from this completely
subjective and unscientific observation has sprung the assumption that there
is always a placebo effect — in all conditions. And so we drill holes into
people’s skulls, when we don’t know whether we need to or not, and
no-one questions the need. Beware
the dreaded placebo effect, my son.
"Beware
the Jabberwock, my son! He
took his vorpal sword in hand: And
as in uffish thought he stood, One,
two! One, two! And through and through "And
hast thou slain the Jabberwock? ’Twas
brillig, and the slithy toves More essays by Malcolm Kendrick TYPE ll DIABETES ISN’T A DISEASE
What
is a disease? Here are a few definitions, culled from three dictionaries:
Okay,
so that counts as pretty unhelpful. A disease is: an illness, an unhealthy
condition, a failure of health, an impairment of normal functioning. I can
sense a circular discussion arriving. There
was a time when I thought I knew what a disease was. Then I started thinking
about it, and realized that the concept of disease is horribly difficult to
get a handle on. Superficially, it seems relatively simple to define disease,
and this is probably most true when it comes to an infectious ‘disease’.
For here we have an agent, and a set of symptoms and signs caused by that
infection. But even in the case of an infection, what is the disease? Where
is it? If
you get infected with the tuberculin bacillus you may develop TB. But TB can
affect the lungs, the gut, the lymph nodes, bone. The infective agent is the
same in each case, but the disease state can vary enormously. Having TB in
the lungs can lead to coughing up blood, breathlessness — death. TB in the
gut can just sit there dormant, unnoticed. Is TB, therefore, always the same
disease, or several different diseases caused by the same agent? Extending
this thought slightly, if we couldn’t find the infective agent in TB,
would we think that lesions in the gut were the same disease as lesions in
the lungs? I suspect not. We would call TB in the lungs, consumption, and TB
in the guts, bowel nodularity — or something of the sort. What
becomes clearer, when you start thinking about things more deeply, is that,
in general, the process of defining disease starts when doctors find an
abnormality. At this point they usually define the abnormality as the
disease, unless, or until, they find a deeper underlying cause for that
abnormality. Thus high blood pressure of unknown cause becomes essential
hypertension, and hypertension is considered by most doctors to be a disease.
Even though there must be a deeper problem that causes the blood pressure to
be high in the first place. Equally,
if you find a number of interconnected abnormalities clustered together,
these are quite often named as a disease after the doctor who first noticed
the connections, for example: Parkinson’s disease, Addison’s disease,
Graves disease, Cushing’s disease, Hodgkin’s Lymphoma, Fallot’s
tetralogy, etc. None
of these doctors had the faintest idea what the underlying cause might be.
They just said that they had seen patients with this set of abnormalities. I
hereby name this set of symptoms and signs… Kendrick’s’ disease. Well,
it has a ring to it. The most recent example I know of is Gerry Reaven of
Stanford University who noticed a number of interconnected metabolic
abnormalities in patients at high risk of CHD. This was called Reaven’s
syndrome. A syndrome, not a disease — discuss. So
you might ask where has all this has got us.The point I am trying to make
here is that our definition of a disease is actually totally arbitrary. I am
sure that almost everyone believes that they know what a disease is, and
what it is not. But when you try to get a grip on it, you will find the
concept slips away like mercury. Does
it matter at all? Is this not just playing with words, asking ‘how many
Angels can dance on the head of a pin?’ Actually it does matter, rather a
lot. Primarily when we try to treat diseases when we do not know, or
haven’t bothered to define, what it is that we are really trying to treat
— symptom or disease; cause or effect. Which, in a roundabout way, is how
we get back to diabetes. Everyone
I speak to is certain that diabetes is a disease. But what is diabetes? The
Greek root of "diabetes" means "siphon," and the Latin
root, "mellitus," means "honey," referring to the
copious voiding of sweet-tasting urine by the diabetes sufferer. From the
first century a.d. onward, other emotional descriptions of this killer
disease included "sugar sickness," "pissing evile," and
"melting down of flesh and limbs into urine." Actually,
that almost certainly wasn’t type II diabetes they were talking about.
These were descriptions of type I diabetes. What’s the difference? Type I
diabetes happens when the insulin producing cells in the pancreas are
destroyed by an auto-immune process — of unknown origin. With no insulin,
the blood sugar rockets up and sugar starts to leak into the urine. Amongst
other things. Type
II diabetes is primarily caused by resistance to the effects of insulin, or
insulin resistance. Usually, there is enough insulin kicking around, but it
doesn’t work so well, so the blood sugar level rises. The different types
of diabetes have gone through a number of different naming protocols. Type I
used to be called juvenile diabetes, as it tended to start at an early age.
Type II was called adult diabetes, for obvious reasons. Type
I and type II diabetes have also been designated insulin dependent and non
insulin dependent, and type A and type B. There is another terminology
kicking around called Latent Autoimmune Diabetes of Adults (LADA), which
describes adults who end up with auto-immune destruction of insulin
producing cells. There is even another type of diabetes entirely, called
diabetes insipidus. And computer people think it’s difficult to keep up
with the speed of change — pah! In
this discussion, however, something is already happening that you won’t
even have noticed. Something critical. Something that you could stare at for
the rest of your life and never even realize that there was anything wrong
at all. An
underlying assumption is now forming in your mind, actually it has already
formed, and it is this. Diabetes is a disease where the blood sugar level
rises too high. (I am restricting the discussion here to type II diabetes by
the way). Of course that is true. Diabetes is a disease where the
blood sugar level rises too high. But what is the disease? The high
blood sugar level? Or the underlying problem that causes the sugar to get
high in the first place. Tracking
backwards in time for a moment. When all that doctors were able see was the
passing of ‘too much sweet sugar in the urine,’ diabetes was called
diabetes mellitus ‘passing too much sugary urine.’ We know that passing
too much sugar in the urine was a symptom, not a disease, yet we got stuck
with a name that merely described a symptom. We’ve still got it. Next,
it was discovered that in diabetes, the sugar level in the blood was also
very high. So diabetes came to mean a high blood sugar level. It still does.
When Banting Best and Mcleod isolated insulin from the pancreas of cows and
injected it into people with type I diabetes, their blood sugar level went
down, and they recovered. Until the insulin ran out, of course. But
it was never the high blood sugar levels that killed a type I diabetic
patient. In diabetes, you die because insulin is required to switch on the
production of sugar receptors from within cells all around the body - other
than in the brain. With no insulin, no sugar receptors are produced, and no
sugar can be absorbed from the blood. With
no sugar to use for energy, the cells start to metabolise fat, and protein.
One of the residues of fat and protein metabolism are ketone bodies, and
these are acidic. After a while this ‘acidity’ cannot be compensated
for, the diabetic falls into an acidic coma and dies. So
when Banting and Best gave patients insulin they weren’t saving life
because they lowered blood sugar levels, even though they thought they were.
By giving insulin they were allowing cells to manufacture sugar receptors,
absorb and metabolise sugar and clear out the acidity from the blood. The
‘disease’ they were treating was not a high blood sugar level — it was
a lack of insulin. But
because the disease, in diabetes, was a raised blood sugar level, it was
just assumed that it was the lowering of the sugar that was critically
important. And even though everyone now knows that type I diabetics die of
diabetic ketoacidosis, the historical baggage that comes with diabetes has
proven impossible to shift. So
we still define diabetes, the disease, as a high blood sugar level. The
current goal of treatment in type II diabetes is to lower the blood sugar
level. But a raised blood sugar level is always a sign of an underlying
‘disease, whatever that disease may actually be. Can lowering a metabolic
sign really prevent mortality and morbidity? Are we treating a disease when
we lower blood sugar levels? No, we are not. We are lowering blood sugar
levels which is an effect, not a cause. Does
this mean that lowering blood sugar levels is a waste of time…. I didn’t
say that, although the evidence that keeping blood sugar levels under
control provides benefit (in type II diabetes) is proving somewhat elusive.
In fact, some studies appear to show that tight blood sugar control may
actually result in increased mortality. This would be surprising if we were
actually treating a disease. But it is less surprising once you recognize
that you are treating a metabolic sign. I
will try to finish where I started with the statement that type II diabetes
is not a disease. It can’t be because type II diabetes is merely a blood
sugar measurement. A sign, an effect. Not a disease, or a cause. We have
become mesmerized by blood sugar levels — we fight to get them down — we
are happy when the level is lowered. Doctors claim, when the blood sugar
level falls below an arbitrary figure, that the type II diabetes has been
treated, even cured. But what, exactly, have we cured? An annoyingly high
figure on a piece of paper that comes back from the laboratory — or a
disease? More essays by Malcolm Kendrick
IS HEART DISEASE ALL DUE TO BLOOD CLOTS?
Up
to now I have resisted writing about this area, as blood clotting is a
mind-boggling and complicated area of human physiology. In the end, however,
if you want to understand coronary heart disease (CHD), you cannot ignore
the role of the humble blood clot. For
it is now accepted by everyone involved in CHD research that the final event,
the thing that kills you with CHD, is the formation of a blood clot on top
of an atherosclerotic plaque. If the blood clot is big enough to completely
block a critical artery, in a critical area, you will die. In
the last few years medics have become increasingly expert at trying to clear
these potentially fatal clots. Aspirin is the first line of defence, then
the clot busters streptokinase, or tissue plasminogen activator (tPA) are
used. Increasingly,
cardiologists get to work with thin wires and balloons, and stents, to
remove the clot, prize the artery apart, and stick a metal framework to keep
the artery open after unblocking it. New drugs have been developed to keep
the artery patent. This is all great stuff, and many thousands of people who
used to die are now being saved. So
there is no argument from anyone about the final event in CHD. It’s a
blood clot. It is also recognised that blood clots develop over
atherosclerotic plaques on quite a regular basis without causing any
symptoms at all, presumably because they are not big enough to fully block
the artery. However,
in these silent episodes, once the blood clot stabilises it adds to the
plaque size, and can lead to greater narrowing of the artery. In this way,
repeated blood clots forming over an area of existing plaque cause
atherosclerotic plaques to ‘grow’. And if you look at plaques closely,
you can — in many plaques — clearly see bands, with each band indicating
an episode of plaque growth. This
is all agreed upon by almost everyone. And if you were a simple soul, like
me, you might argue that if plaques grow, and eventually kill you due to
clots forming on the artery wall, could this not be how they start in the
first place? Are atherosclerotic plaques not, in fact, just the remnants of
repeated blood clots, which are ‘drawn in’ to the artery wall, in time
turning into a form of scar tissue? If
you did think this, you wouldn’t be the first. This hypothesis was
initially proposed by Karl Von Rokitansky in 1852. Although supporters of
Rudolf Virchow may argue that he said it first. Unfortunately, therefore, I
can hardly claim that this idea is either new, or mine. Can
it really be that simple? Surely there must be something wrong with the
hypothesis that atherosclerotic plaques are the remnants of repeated blood
clots? Where does this idea break down? I could say, don’t ask me, I
happen to believe it’s true. But I will attempt to be a little more
objective than this. The
key point of objection is that, whilst you can see how blood clots can form
over a ‘damaged’ artery wall, it is very difficult to see how they form
over a healthy artery wall. After all, a critical function, perhaps the
critical function of the lining of the artery (the endothelium) is to
prevent blood clots from forming. So how can this process actually start? A
good point from my learned friend. But
I put it to you, members of the jury, that every ‘factor’ that has been
identified as increasing the risk of CHD, has clearly identifiable
pro-coagulant activity. Equally, every factor that has been identified as
reducing the risk of CHD has clearly identifiable anti-coagulant activity.
Which, I would vouchsafe, is pretty heavyweight proof. Is
this really true? Well,
yes. But you have to understand that there are three interconnected factors
at play here that can cause a clot to form over the artery wall. Factor one,
is ‘damage’ to the endothelium. Once damaged, the endothelium stops
acting as a non-stick anti-coagulant surface. Indeed, if the endothelium is
stripped away, it exposes the middle layer of the artery, the media, to the
blood, and the media releases the most powerful pro-clotting factor known to
man: Factor VII, the ‘extrinsic’ factor. The
second factor is how pro-coagulant the blood is itself. There are a
multitude of clotting factors in the blood. Some of which you may have heard
of, such as factor XIII - the one that is missing in haemophiliacs; some of
which you probably haven’t heard of e.g. Von Willibrand factor. Increase a
pro-coagulant factor, and you increase the chance of clots forming. The
third factor is the structure of the blood clot itself. Some clots are
wobbly and weak; others are very tough, and difficult to break up. For
example, incorporated into all blood clots is a substance called plasminogen.
This is an enzyme which, when activated, chops the clot into pieces. (Which
is why tPA — tissue plasminogen activator - is given to people having a
heart attack) However, if you have a high level of Plasminogen activator
inhibitor — 1 (PAI — 1) in the blood, plasminogen is less effective at
breaking the clot up. So,
you have to look at three basic factors:
Bearing
this in mind, I think it is interesting to run through a few factors known
to alter the risk of CHD, and see how they fit: Smoking:
Smoking
creates free-radicals in the blood, these reduce nitric oxide (NO) synthesis
in the endothelium, and NO is the single most powerful anti-coagulant factor
in the body. Smoking also has pro-coagulant effects in the blood; it raises
fibrinogen levels. It also has endothelium damaging effects. So, if you want
to avoid CHD… STOP SMOKING! Drinking: Ethanol,
in moderate doses, reduces free-radical synthesis, reduces clotting factors,
such as fibrinogen, and reduces the blood clot toughness. However, excess
alcohol consumption creates rebound platelet stickiness (platelets are
hugely important in blood-clotting). Moderate drinking protects against CHD,
heavy drinking is a risk factor. Diabetes: A
high blood sugar level leads to increased free-radical synthesis, see above.
A high blood sugar, independent of its effects on NO synthesis, also causes
endothelial ‘damage.’ Haemophilia:
Not
surprisingly, haemophilia reduces blood coagulability. Haemophilia also
reduces the absolute risk of CHD by 80%. Statins:
Statins
have strong anti-coagulant effects, they stabilise plaques and increase NO
synthesis. Aspirin:
Aspirin
reduces the stickiness of platelets (see alcohol). Platelet aggregation is
the first step in blood clotting. Omega-3
fatty acids Omega-3
fatty acids have strong anti-coagulant effects in the blood Stress Physical,
or psychological stress causes the release of the stress hormones: cortisol,
adrenaline, growth hormone and glucagon. These hormones all increase blood
coagulabiltiy, raise the blood sugar level (see above), and ‘damage’ the
endothelium. Raised
blood pressure
I
am a little ambivalent about this risk factor. I am unconvinced that a
raised blood pressure really is a ‘cause’ of plaque development. However,
it is possible to see how high pressure, and turbulent blood flow, could
strip away a layer of endothelium, exposing the blood to the media, and thus
factor VII, thus stimulating a blood clot to form. It is certainly true that
plaques don’t form in low pressure blood vessels (e.g. veins). However,
the clinical trials on blood pressure lowering are very unconvincing when it
comes to a correlation between the degree of blood pressure lowering and the
prevention of CHD. HDL
HDL
has strong anti-coagulant effects LDL
(Oxidised LDL)
This
is a complex pathway. When platelets start to stick together, they release
free radicals. Free radicals oxidise LDL. Oxidised LDL is a powerful blood
clotting factor. LDL is also incorporated into the blood clot as it forms,
and provides a ‘lipid’ surface (along with VLDL) for the construction of
fibrin. Fibrin is the hugely strong protein strand that binds a clot
together and makes it ‘tough.’ Frankly,
I think that’s enough. If you wish to, it is possible to link every single
factor known to have an impact on CHD rates to one of three effects:
endothelial damage, blood coagulation, or toughness of the clot. If you
don’t affect any of these three things, then you have no effect on CHD
rates. If you manage to impact all three, then it’s time to increase the
life insurance. (On a positive note you won’t need a big pension fund). So,
if it’s that simple, then why have you never heard of this before? Now
that is another story altogether. But if enough people think I am making all
of this up, then I will provide a series of references from prestigious
journals to support every single fact that I have presented. To
my mind, the answer as to the underlying cause(s) of CHD is not only ‘out
there,’ it has been staring everyone in the face for the past fifty years.
Perhaps it is too obvious for anyone to see it. To
quote a riddle that my son came home with the other day: What
is greater than God Once
you know the answer you cannot believe that you couldn’t see it
straightaway. More essays by Malcolm Kendrick IS IT ALL A BIG FAT LIE?
A
number of people have written, asking me to comment on the Gary
Taubes-Michael Fumento battle. What do I think, what is true? Over
on this side of the pond, most people have never heard of Taubes or Fumento,
or Dr Aktins. But the pro and anti-fat battle seems to be raging in the
colonies, with people hurling data of mass destruction at each other with
great vigour. As usual, I find that ‘truth is the first victim in any war.’ So
what is the truth? Who is right, the pro or anti-fat lobby? The question, in
the case of Taubes vs. Fumento appears to split in two. Does eating fat or
carbohydrates make you fat? Secondly, does eating fat or carbohydrate give
you heart disease? For the sake of brevity, I’m not going to deal with the
second question here. Unfortunately,
any discussion of food and eating carries a huge emotional baggage. It
becomes wrapped up with issues such as vegetarianism, greed, guilt, sin,
pleasure. You name it and Freud would have had a field day. After sex, food
has probably screwed up more ids, egos and superegos than anything else. So,
the context is not exactly ideal for any rational discussions. In this area,
people tend to believe the facts that they like, and discount the facts that
they do not. And when you study diet and health, you can find hundreds of
studies to support almost any position that you care to take. Anyway,
with regard to eating fat and getting fat: Where to start? Perhaps with some
facts that are accepted as absolutely true by almost everyone. I should warn
you that there are not too many of these, and you will probably disagree
with at least one of them. Fact
one, fat contains twice as many calories as carbohydrates, weight for weight.
So if you ate one hundred grams of fat you will be eating twice as many
calories as if you ate one hundred grams of carbohydrates. Fact
two, the body has two ways of getting rid of energy. Physical work and heat
generation. We cannot, as far as I am aware, radiate energy through
electromagnatism, light, microwaves, or any other high intensity radiation.
Nor does the body have any way of shedding excess fat through excretion by
the kidneys, bile, faeces, sweat or saliva. In short, once you have stored
energy as fat it has only two ways of being used up. Fact
three, whenever anyone has studied this area closely, i.e. putting people
into a closed environment and accurately measuring calorific input and
output, the two match. Big surprise? Fact
four, there is no such thing as a ‘naturally’ high or low metabolic rate.
Thin people eat less than fat people — with three provisos. The first
proviso is that exercise burns calories. I read that in the Tour de France
cycle race, the competitors use about twelve thousand calories a day. And
people who walk across the South Pole use about six to eight thousand
calories a day. So, exercise can keep you thin. The
second proviso is that a seven-foot tall male will naturally burn up more
calories than a five-foot woman. So size and sex have an impact on the
number of calories needed for energy equilibrium. The third proviso is that
some diseases can affect the metabolism, such as thyroid disease. What
else is known for sure….. well, nothing very much. Does the weight of
evidence support the hypothesis that eating fat makes you fat? No. Does the
weight of evidence support the hypothesis that eating carbohydrates makes
you fat? No. What of Dr Atkins and his marvellous diet? Does it work? I’m
sure it does. Any calorie restricted diet works, so long as you stick to it.
Does it work in the long term? I’m sure it does if you stick to it in the
long term. Gosh,
how boring. If you want to lose weight, eat less or exercise more. What did
you want to hear? That fat has some magical, weight losing, second law of
thermodynamic altering properties. Those things could possibly happen in
another Universe, but not this one. A calorie is a calorie is a calorie.
They don’t just disappear — unless you use liposuction. End
of discussion? Perhaps. But there is a more interesting question lurking in
the depths, which I shall call the satiety question. Does eating too many
carbohydrates create a metabolic situation that results in people eating
more than they would if they ate something else, such as fat? The
hypothesis underlying this question is fairly simple and it goes something
like this: If you eat sugar, this will be absorbed very rapidly, causing a
sudden ‘kick’ in blood sugar levels. This triggers insulin release and
within a short period of time all the sugar will have ‘gone’ from the
bloodstream. However,
with the insulin still high, your blood sugar level drops, causing a sense
of hunger and a need to eat again, very quickly. So you enter a vicious
cycle of hunger, carb intake, blood sugar spike, insulin release, blood
sugar fall, hunger. Or something of the sort. On
the other hand, if you ate the same amount of calories as fat, this would
take much longer to absorb, it would not cause the sudden surge in insulin,
and ‘food’ would hang around for longer in the bloodstream. You would
not get ‘rebound’ hypoglycaemia and rapid return of hunger. A
reasonable hypothesis, therefore, is that carbohydrates may create a
‘hunger’ cycle, and cause people to eat much more, thus getting fatter.
But although this is a reasonable hypothesis, I don’t think there is
strong proof to support it. My own difficulty with this hypothesis is that
most people don’t seem to eat because they are hungry. In
fact, there seems an almost perfect dislocation between true hunger and
eating, in our rich Western societies. I’m never hungry on a plane, but a
member of the cabin crew inevitably slaps food in front of me, and I end up
eating some of it. Eating, and the reasons why people eat, is much more
complex than ‘I eat when I am hungry, and I don’t eat when I am not
hungry.’ If people did act like this, I suspect that obesity would never
have been heard of. People would travel miles to see ‘The amazing man who
stores fat around his waist…. Gasp.’ But
we have managed to completely screw ourselves up when it comes to food. When
we are young, for example, we are praised for clearing our plate. I don’t
recall any parent stating, ‘Thank Goodness you stopped eating when you
were full, and did not keep stuffing it down just to please me.’ Instead,
I have memories such as the one where I watched a fellow inmate at my school
being forced to finish food by a terrible, unbending schoolmaster, unaware
that another boy had spat in it — or maybe he was aware. Most unpleasant.
Oh yes, over the years we have managed to turn food into a battleground. Anorexia,
bulimina, diets, guilt, anger, fear, praise, ridicule, pain, sickness…..
Amongst all of this horrible mess how the hell do you think you are going to
prove anything? I just finished reading Gary Taubes reply to Michael Fumento.
(See other articles in this debate section) And the best quote came from
Ruby Liebel of Columbia discussing research on weight gain and weight loss: ‘When
I interviewed Leibel, who is now at Columbia University, he capped our
conversation this way: "if you do feel you understand this," he
said, "it will probably indicate that you've lost your mind."’ It
partly boils down to this, I think. Whilst having a big brain carries
certain advantages for us humans, it also creates great difficulties. For
our brains are immensely powerful, and they release powerful hormones into
the system. When our brains get upset, they screw up the entire metabolic
system by firing off all sorts of neurohormonal messages. With
this huge brainpower at our disposal, we can easily over-ride hunger, or a
sense of being full. We can eat for social reasons, the need to be liked,
the fear of criticism. We don’t eat if we fear being fat, losing control,
being laughed at. We eat when we are stressed to calm ourselves, even though
every hormone in the body is screaming at us to stop. When we diet we
don’t really feel hunger, we just resent being denied things we want. We
eat to bring pleasure to our brains, and the hell with what our body wants
to do. So
you want some simple answer? Eating fat makes you thin? Eating fat makes you
fat? Not a hope. There are a few too many variables here that, as yet, are
not measurable by medical science. To quote Albert Einstein ‘Not
everything that matters can be measured, and not everything that can be
measured matters.’ So,
does eating fat make you fat? I don’t think so. I think that driving a car
rather than walking a hundred yards makes you fat. I think that the easy
availability of food, especially fast food, makes you fat. I think that
connecting food and eating with concepts of good and bad, pleasure and guilt,
makes you fat. I think that disconnecting our minds from the messages that
our bodies are desperately trying to send us makes us fat. Finally,
I also think that nature quite likes us humans to store fat in times of
plenty, so that we don’t starve to death if the food supply dries up. Some
animals are designed to get fat, others are not. We are. If we weren’t, we
wouldn’t. Try fattening up a chicken some time — not possible. But never mind, someone somewhere is going to invent a pill that makes us all thin. More essays by Malcolm Kendrick
OKAY, DO YOU KNOW WHAT A "FAT" IS?
I
received an overwhelming response to my little
primer on lipoproteins,
so I thought I should explain a little more about fats. Excuse my diagrams,
I got them all from the internet, so they have no overall design template,
but I hope that I can keep things clear. A
fat has the basic structure shown below (Fig 1). (Fig
1) My nameless fat
All
fats are, basically, a chain of carbon atoms of varying length, where the
carbon atoms are attached exclusively to hydrogen atoms, apart from the
group at the end — COOH - called a carboxyl group. The carboxyl group is
what defines fats as an acid, or fatty acid. So a fatty acid and a fat are
actually the same thing. The terms are interchanged at will. That one caused
me endless confusion. You
may have noticed that my nameless fat has a gap at the bottom, where no
hydrogen (H) atoms are attached. And there is a double bond between the
carbons with the missing hydrogens. This means that the carbon atoms at
either end of the double bond are not ‘saturated’ with hydrogen. So this
fat is ‘unsaturated.’ The
other thing about my nameless fat is that there is only one double bond, so
this fat would be referred to as a mono-unsaturated fat. Monounsaturated =
one double bond, or two hydrogens missing. If there is more than one double
bond, the fat is referred to as poly-unsaturated. Clearly,
therefore, a saturated fat is one with no double bonds, and no hydrogens
missing. It is fully saturated with hydrogen. You can see how this
nomenclature works in Fig 2. In
real-life you can tell if a fat is saturated primarily because it is solid
at room temperature. Monounsaturated fats and polyunsaturated fats are
usually liquid are room temperature. Unless you live in Siberia, of course. Fig 2
So,
how come you can spread margarine, it being polyunsaturated and all? Because
in the 1930s a very clever chemist learned how to nail extra hydrogens to
unsaturated fats using a process known as ‘hydrogenation,’ and thus was
born margarine — hoorah. Artificially hydrogenated fats are often called
trans-fats, or trans fatty-acids. These types are fat are not really found
in nature at all. As
a slight aside, forget GM foods, margarine is as alien as it gets. Even if
you call it Flora and paint pictures of lovely flowers around the tub, and
get highly paid athletes to promote its health giving wonders.
‘Flora….’ (is this just a UK name?). Sorry, start again with my
strap-line (fade-in Beethoven’s pastoral symphony). ‘Flora, as natural
as platinum catalysed hydrocarbon cracking itself.’ That, by the way, is
what we in the UK call a joke. Just in case the Flora lawyers try to sue me. So,
now you know the difference between a saturated and unsaturated fat. And
pretty unexciting it is too. How come it seems so difficult? Because people
start using terms like alpha-linoleic, and stearic and Omega-3, and
cis-bonds and uncle Tom Cobbly and all. All
of this is really just a form of nomenclature used by chemists (to confuse
us poor laypeople). However, to keep it simple, if the double bond in a fat
is three along from the ‘Omega’ end of the fat, which is the opposite
end from the carboxyl group, the fat is then called an Omega 3 fatty acid.
If the double bond is six along, it is an Omega 6. And that’s the
difference between an Omega 3 and an Omega 6 fatty acid. Wow — hold onto
your seat — is this exciting or what. Where
do the other names come from e.g. stearic, and linoleic, and palmatic etc?
Generally, these names are taken from the source of the fat. So palmatic
acid comes from Palm oil/fat. Linoleic comes from linoleum (only joking —
it’s vice-versa). These types of fat/oil are defined primarily by the
length of the carbon chain. Linoleic acid, for example, has eighteen carbon
atoms. Thus,
fats are named according to a few different variables. Where they come from,
palmatic, coconut (and this also defines the number of carbon atoms),
whether they are saturated, or unsaturated, and where the double bond, or
bonds, sit. The other significant bit of nomenclature is whether or not the
double bond is cis, or trans. To explain. Usually,
in natural fats, the hydrogen atoms sit on the same side of the double bond,
causing a ‘kink’ in the chain. See diagram 3. when you get this
‘same-side’ hydrogen structure, the bond is known as a cis bond. If the
hydrogens are spread either side it is known as trans. Because
cis bonds have both the hydrogen on the same side, they tend to kink,
causing the chain to bend. This bendyness allows the fat to wiggle around
more, and so the fat is fluid. A saturated fat has no kinks, no bendiness,
and thus remains solid. Trans bonds are also less wiggly than cis bonds, so
the fat is more solid, but no too solid. Which is why margarine can spread
‘straight from the fridge,’ or in my case, straight into the dustbin. Diagram 3
And
that’s about all you need to know about fats — or fatty acids. There are
other ‘naming’ protocols, but they don’t really have much relevance to
non-specialist audiences. But
please keep one thing in mind - within the context of heart disease. The
only real connection between fats and cholesterol is that, as they are
insoluble in water, they have to be transported around inside lipoproteins.
You don’t make cholesterol from saturated fats, or any other sort of fat,
or vice-versa. So,
why do people keep telling you that excess saturated fat consumption raises
your Cholesterol level? Because this has become an article of faith. It is
not susceptible to reason, logic or facts. Metabolically speaking, there is
no connection between these two substances at all. They just happen to sit
in the same lipoproteins. (And it’s the lipoprotein level you’re
interested anyway — you don’t actually have a cholesterol level) Equally,
why do unsaturated fats lower your cholesterol level? They don’t. How
could they? Fats, saturated or otherwise, ARE NOT CONNECTED TO CHOLESTEROL
METABOLISM. You might as well argue that eating excess protein will raise
your blood sugar level. For a graphical illustration of the differences
between fats and cholesterol. See diagram 4. DIAGRAM
4 A
SATURATED FAT CHOLESTEROL
So now you know what a saturated fat is, and what an Omega 3 fatty acid is, and what cholesterol is. I hope you will now find what I found. Once you understand this stuff a bit better, you can’t imagine why anyone ever thought that saturated fat in the diet had an impact on cholesterol levels in the first place. There is just no connection.
I
have written a few columns
on heart disease for Red Flags and the response has been very positive.
However, there is a major problem that emerges quite clearly from e-mails
that I get back. The problem is that there is an enormous level of confusion
about the whole area of cholesterol, lipids, lipoproteins, fats etc. So I
thought I should provide a simple primer on this area, as it makes debate
and discussion a lot easier. Before
getting into the area, I must admit that I have a great deal of sympathy
with the confusion. When I first started looking at the diet-heart/cholesterol
hypothesis I found the science to be almost totally incomprehensible, and
much of this is due to, what I refer to, as terminological inexactitude. To
provide a couple of simple examples. A high level of low density lipoprotein
(LDL) in the blood is usually referred to as a high cholesterol level.
A high level of very low density lipoprotein (VLDL) in the blood is usually
referred to as a high triglyceride level. Frankly this is nuts, as
LDL and VLDL contain both triglycerides and cholesterol - and neither
triglycerides or cholesterol float free in the blood. Let’s
try another example. An LDL with a protein attached to it called
apolipoprotein b-100 is called LDL. LDL with a protein attached to it called
apolipoprotein (a) is called Lipoprotein (a). Or Lp(a)…. I sense confusion
arising. So,
let’s start at the very beginning, it’s a very good place to start.
Point number one. cholesterol is not a fat; it is called many different
things, even an alcohol, but one thing it is not, is a fat, or a fatty acid.
(Fats and fatty acids are the same thing, by the way). Nor can you make
cholesterol from fats. Cholesterol
starts life as a chemical called Acetyl coenzyme a. A relatively
ubiquitous building block that is used to make all sorts of things that the
body needs. The vast majority of cholesterol in your body is synthesised by
the liver from Acetyl coenzyme a. You only get about a quarter of
your cholesterol from dietary sources. Point
number two: Triglycerides are three fat molecules stuck to a Glycerol
molecule - which is where the tri and the glyceride come from.
Although the fat part seems to have gone missing in the nomenclature. Most
fats are transported around the body and stored as triglycerides. When
you eat cholesterol and/or fat, they are absorbed by the gut. But neither
fat/triglyceride, nor cholesterol can be dissolved in blood - they are
insoluble in water. So, they have to be wrapped up in a sphere known as a
lipoprotein in order to transport them out of the gut. Point
three: Lipoproteins come in many sizes. The biggest is a chylomicron and the
smallest is a high density lipoprotein (HDL). If a chylomicron were the size
of a football (soccer ball), a VLDL would be the size of a baseball, an LDL
would be the size of a golf ball, and an HDL the size of a pea, perhaps even
a petit pois. All
lipoproteins contain cholesterol and triglyceride - in varying proportions.
The basic function of a lipoprotein is to carry triglycerides from the gut,
or the liver, to fat cells, where the triglyceride is then stored and used
for energy when needed - in situations such as pressing the remote control
for the television, or chewing a hamburger. Lipoproteins
also transport cholesterol and triglycerides to the liver. When a
chylomicron reaches the liver, from the gut, it is grabbed, absorbed, and
then smashed to pieces. The liver then reconstructs the component parts into
VLDLs and sends them out into the bloodstream with an apolipoprotein b-100
protein stuck to the side. As
a VLDL travels around the body, fat cells snatch at it, chop bits off and it
gets smaller and smaller, turning first into an intermediate density
lipoprotein (IDL), then a low density lipoprotein (LDL). Once the
lipoprotein has reached LDL size, it is either re-absorbed by the liver and
re-used, or it is absorbed by other cells around the body that are in need
of cholesterol. The
reason why LDL can be absorbed is all to do with the apolipoprotein b-100.
This is the protein ‘key’ that the cells recognise. It is the key that
fits exactly into the LDL receptor on the cell wall. Once the b-100 molecule
locks to the receptor, the receptor closes around the LDL, draws it into the
cell where the LDL is broken down into its component parts. And
what of High Density Lipoprotein (HDL)? HDL is not part of the same
metabolic ‘loop’ as the other forms of lipoproteins. It is made
separately, and appears to act as a cholesterol mop, scavenging loose
cholesterol from broken down cells and suchlike, and transporting it back to
the liver. Which is why it is often called ‘good’ cholesterol. It is
called this even though HDL isn’t cholesterol, and cannot possibly have
any effect on removing cholesterol deposits from arterial walls. In short,
it is neither cholesterol, nor good. Apart from that it is a magnificently
accurate form of nomenclature. Anyway.
In short, lipoproteins are the ‘taxis’ that are used to transport
insoluble cholesterol and triglyercides around the body. Apart from HDL,
lipoproteins start big, as chylomicrons, and gradually get smaller as they
lose triglyceride. The VLDLs, produced by the liver get smaller and smaller
until they become LDLs. At which point they are reabsorbed into the liver,
or other cells. What
then, is the cholesterol level? Well,
it should be obvious by now that the cholesterol level doesn’t actually
exist. For there is no cholesterol free in your bloodstream. You can have a
level of LDL, or VLDL, or chylomicrons, but you can’t have a level of
cholesterol. And so all measurements of ‘cholesterol’ are actually
measurement of lipoproteins - which is where most of the terminological
confusion arises. Thus,
when someone uses the term ‘total’ cholesterol, what they mean is the
level of LDL, plus HDL, plus a few other lipoproteins e.g. Lipoprotein (a)
and/or some intermediate density lipoproteins that aren’t quite LDLs, but
get mixed up in the process. When
the term LDL/cholesterol, or ‘bad’ cholesterol level is used, this
refers only to the level of LDL. This is usually about two thirds the level
of ‘total’ cholesterol. Other laboratories will tell you both the LDL
and HDL (good cholesterol) level, and give you the proportion of LDL to HDL.
With a ratio greater than three seen as ‘bad’ and a ratio less than
three as ‘good.’ This can be presented as LDL:HDL 3.2:1, or whatever. Some
people think the level of VLDL is important, and they will give you this
measurement as well. But they will call it the level of triglyceride. I think that is enough for one article. I hope that you find it a helpful dash through the nomenclature used in this area. If I get some positive feedback I could explain the difference between saturated fats and unsaturated fats, and what the terms Omega 3 and Omega 6 actually mean, and a few other things as well.
HIGH
BLOOD PRESSURE: IT’S A SYMPTOM, NOT A DISEASE, STUPID!
Let’s
suppose that one day you went to the doctor and she decided to take your
temperature, just to see what it was. To your surprise it was two degrees
higher than normal. As we all know, a high temperature is associated with a
higher than normal level of mortality, so the doctor decided to use a drug
to get your temperature down, along with advice to wear less clothes and
take cold baths. Time
passes and you have been on this drug for five years. The baths and chilly
walks are getting to be a bit of a pain. On the bright side, at least the
temperature is back to normal. I
think you would agree that such a scenario is, quite frankly, nuts. Yet,
every day, thousands of people are found to have high blood pressure, and
put on blood pressure lowering drugs on pretty much the same basis. The
logic, after all, is the same. 1.
People with high blood pressure are more likely to die from CHD 2.
Therefore a high blood pressure causes CHD 3.
Therefore if you lower the blood pressure you will reduce the rate of
CHD 4.
So, take a blood pressure lowering drug for the rest of your life I
suppose that most people believe that it must have been proven by now that
blood pressure lowering does reduce the rate of CHD, rendering the example
of a high temperature somewhat pointless. Well, I am going to quote you
quite a long passage from the European Heart Journal, issue 20, October
2000. Please read it carefully, for it is actually quite stunning. ‘It
is widely believed that randomised trials have proved that lowering blood
pressure is beneficial. Actually, that is not true. All antihypertensive
drugs have profound effects on the cardiovascular system, aside from their
haemodynamic (blood pressure lowering) effect. How much, if any, of the
observed risk reduction can be ascribed to the reduction in pressure and how
much to the direct action of the drug on the cardiovascular system?
Motivated by the belief in the linear relationship of risk to pressure, many
automatically attribute the risk reduction to the pressure reduction,
ignoring the direct action of the drugs on the target outcomes. But results
of a multitude of clinical trials make it clear that such a simplistic view
cannot be true. In fact, evidence is mounting (especially from the newer
trials) that it is the direct effects that are producing most, if not all,
or the benefit and that the accompanying blood pressure reduction may be
just an inconsequential side effect.’ Port S et al. In
short, there is no evidence whatsoever that lowering blood pressure has any
effect on CHD. As they authors of the paper further state: ‘ALLHAT
(A major blood pressure lowering trial) showed a dramatic difference between
alpha blockers and diuretics, with essentially no difference in blood
pressure between the treatment and control groups.’ Quelle
surprise? Not really, after all, the underlying hypothesis that blood
pressure causes CHD was always nonsense. After all, how could a high blood
pressure make atherosclerotic plaques form? Well, you can create a
convoluted argument involving endothelial damage, but you would struggle to
create a clear cut case. On
the other hand, it is very much more simple to see how an atherosclerotic
plaque, by narrowing an artery supplying blood to a vital organ, can trigger
the heart to pump harder, thus overcoming the narrowing in the artery by
increasing the blood pressure. Perhaps
it is time for a rewind. What is a high blood pressure, and what could cause
it? In about ten per cent of cases there is a clearly established cause for
high blood pressure. Conditions such as renal artery stenosis, or
hyperthyroidism, or kidney problems. If these are treated, the blood
pressure drops back to normal. However,
in about ninety per cent of cases when the blood pressure is raised, no
cause can be found. At which point the medical profession, rather than using
the somewhat pathetic sounding term ‘Raised blood pressure of no known
cause,’ decided to rename the condition Essential Hypertension. You’ve
got to admit, this sounds a great deal more scientific and ‘disease
like.’ In fact it sounds so impressive that Essential Hypertension has
managed the transformation from ‘symptomless medical sign’ to a real
disease, one that needs to be treated. Let’s
examine the logic in use here. One day, for no known reason, your body
decides that the blood pressure needs to be raised. So your heart pumps
harder, or your arteries decide to contract, or both. This has the desired
effect of raising the blood pressure to a point where it can cause damage.
It can lead to strokes, heart failure, kidney failure, etc. Undeterred
by the damage that this raised blood pressure is causing, the body continues
for day after day, month after month, year after year, to keep the pressure
up. Eventually your heart can’t carry on any more, so it starts to pack
in, you develop heart failure, and within about five years you are dead. There
is just one teensy little thing missing from this model. A cause. Why does
the pressure suddenly rise? One thing is for sure, the body does nothing
without a cause, especially if the effect is to damage health. So we need to
ask a deeper level question. What could cause the blood pressure to rise? In
order to understand this, you need only to the grasp the exceedingly simple
concept that the pressure of liquid flowing through a pipe is a function of
two variables. The first variable is the rate of flow of the liquid; the
second is the diameter of the pipe. If you want to increase the pressure you
must pump more fluid, or narrow the pipe. Therefore,
if your blood pressure goes up, for no known reason, one of two basic things
is happening. 1.
The heart is pumping harder 2.
The diameter of the arteries has narrowed (causing the heart to pump
harder to keep the blood flow the same) Things
that make your heart pump harder would include: anxiety, exercise,
stimulants e.g. coffee. Things that narrow your arteries would be……? Dum
de dum, let me think. Oh
yes. An atherosclerotic plaque (the underlying cause of CHD) would narrow an
artery. Therefore, a probable cause of high blood pressure is the presence
of CHD. Thus,
ergo etc. a high blood pressure is not a cause of CHD. Instead CHD is a
cause of high blood pressure. So yet again gentle reader, as with raised
cholesterol levels and CHD, we see another rather grisly example of the
medical profession grasping the wrong end of the stick and desperately
trying to cure a disease (CHD) by sweeping a symptom of that disease (high
blood pressure) under the carpet. No big surprise, it doesn’t actually
work. Does
this all seem incredibly basic? It should, because it is. So, whilst blood
pressure lowering may have some effect on preventing strokes, heart failure
and other pressure related problems, it has no effect on reducing death from
heart attacks. After all, how could it? Some
of you may have seen research reported in the New Scientist magazine which
established quite clearly that most scientific researchers don’t bother to
read the full papers that they use for references. In fact, most of them
just copy and paste the list of references used in other papers. This
may seem a somewhat arcane issue, removed at two steps from real life. I can
sense a collective ‘so what?’ resonating round the world on this issue.
But please pay attention, because this fact is VITALLY IMPORTANT! And it
explains much about the treatment paradigm for high blood pressure. In
medical science, many measurements are imprecise. A blood pressure taken at
ten in the morning may have changed five minutes later. The doctor may have
put the blood pressure cuff on in a slightly different way, whatever. So,
when you start drawing a graph of blood pressure measurements taken over
time vs. the rate of death, in different groups of people, it does not have
precise cut-off points. It may look more like someone has fired blunderbuss
at a piece of graph paper.
However,
if you are really clever and understand mathematics and calculus, and
suchlike, you can draw a perfect line through that mass of dots. This can be
called a ‘linear logistic model.’ (My line is just a random guess by the
way)
However,
to quote the European Heart Journal: ‘Before one can have confidence that
the linearity correctly reflects the behaviour of the data, and is not just
an artefact of the model, it is necessary to carefully examine the data in
relation to the proposed model.’ In plain English, stop guessing. Although
guessing does look a lot more impressive when you use terms such as Cox
model and double-tailed chi-squared, etc. which no one understands. So,
what does this all have to do with the price of beans? I
have two strands to my discussion so far. Strand one: most researchers never
bother to read the papers they quote; at most they manage to read the
abstract. Stand two: statistical models used to look at blood pressure vs.
mortality are all based on the supposition that ‘the relation of blood
pressure to risk of death is continuous graded and strong, and there is no
evidence of a threshold.’ Now,
where did this supposition first come from? Our old friend Framingham, the
world’s longest and most detailed study of the relationship between
various ‘risk factors’ and death from heart disease. Researchers looking
at the Framingham data started the ‘linear and continuous’ ball rolling,
and, ever since, everyone has decided to use the same methodology. A
statistical methodology which implies that the lower the blood pressure the
better, and there is no lower limit. No
one questions this methodology; in fact it has been quoted in so many papers
over the years that it would appear to have been proven beyond the shadow of
a doubt. But of course, the reason why it is now quoted so often is that
paper after paper has quoted from other papers that have all shown this
linear regressive model to be true. A process of error reinforcing error. To
give a more concrete example of how this happens. I write a paper which
states that ‘the relation of blood pressure to risk of death is continuous
graded and strong.’ Someone else comes along and quotes that paper,
without bothering to look at the methodology or results. So now I have two
papers making the same statement. Then,
along comes researcher B, who is looking for papers on blood pressure and
mortality. He sees two papers with the same self-reinforcing statement on
it, and quotes them. Now I have three papers making the same statement. How
long before there are one hundred, two hundred, a thousand papers? You
think this number may be an exaggeration, but Simkin and Roychowdury (who
looked at the issue of misreporting) found that mis-citations can occur many
thousands of times. To quote the New Scientist article again: ‘To
find out how common this (misreporting) is, Simkin and Roychowdhury looked
at citation data for a famous 1973 paper on the structure of two-dimensional
crystals. They found it had been cited in other papers 4300 times.’ And
the errors this leads to are not specific to two-dimensional crystals: ‘The
problem is not specific to this paper, the researchers say. Similar patterns
of errors cropped up in a dozen other high-profile papers they studied. The
trouble is that researchers trust other scientists to repeat the key message
of a paper correctly. This means that when misconceptions take root, they
spread like weeds.’ It
should be clear by now, where I am heading. Someone,
somewhere, decided that there is a continuous linear relationship between
death and blood pressure. They used a statistical method to establish this,
and ever since everyone has used the same model. So there are now thousands
and thousands of papers out there ‘proving’ this paradigm to be true. In
fact, if you wrote a paper on the treatment of high blood pressure using
another model it would almost certainly be rejected on the basis that the
linear relationship model was the established, and correct, model, so yours
must be wrong. There
is just one teensy, weensy, little problem here. When you actually decide to
look at the data - it disproves the model. ‘Shockingly
we have found that the Framingham data in no way supported the current
paradigm to which they gave birth. In fact, these data actually
statistically reject the linear model. This fact has major consequences.
Statistical theory now tells us that the paradigm MUST be false..’ EHJ
2000 21, 1635 - 1638 I
didn’t add the italics or capital letters. The Authors put them in - the
paradigm MUST be false. Normally, in clinical papers, people state things
very calmly, e.g. ‘the data suggests an association between.’ So to see
a statement such as the paradigm MUST be false is very strong stuff. So
what is really being stated here that is so important? I
will use an analogy to try to make the point. If you chose to live in the
Himalayas you may find yourself twelve thousand feet above sea level. Most
people can cope with this height, and it has very little impact on your
health or life expectancy. Go up a few thousand feet and everyone dies. The
exact ‘death zone’ height varies from person to person. The
fact that you die at sixteen thousand feet, however, does not mean that any
altitude above sea level is harmful. What it means is that, at a certain
level, your body cannot cope any more and the systems start to break down. Yet,
with blood pressure, any rise represents a risk - according to the linear
model. There is no ‘death zone’ no cut-off point. According to this
logic, even if you have a ‘normal’ blood pressure, it would be better if
you could get it lower. And believe me, papers have been written stating
this. But,
anyone with half a brain can see that a model with a ‘cut point’ is much
more likely to be correct. Is it really likely that a 5-15mmHg rise in blood
pressure will cause problems? According to the linear model, the answer is
yes. But, as we have seen, the data doesn’t actually support a linear
model, and logic would also dictate that at a certain point - which has, in
reality, never been defined - a raised blood pressure creates problems.
Below that point it may be a bit high, but frankly it’s nothing to worry
about. What
is that point…. I don’t know. But I would guess it is something like a
systolic of 160 - 180. However, the medical profession, with its ever
present desire to squeeze all patients into a little box called ‘normal’
is inexorably bringing down the level at which treatment is needed. I have
seen calls to get everyone to a level of 120/70 (the level considered
‘normal’). The WHO has set the limit at 130/85. Already in diabetes the
recommended level is 120/85. Why are they trying to achieve this? On the basis of a model made up years ago which, due to sloppy research, has become accepted fact. On the basis of a model which, if you examine it properly, MUST be wrong. Try explaining this to your local, friendly doctor, you will get the same reaction that I always do. ‘Don’t talk rubbish, it has been proved that you should lower the blood pressure as much as possible.’ TREATING
HIGH BLOOD PRESSURE: WHAT CAN YOU BELIEVE?
I
have written about hypertension a couple of times. So I thought that I
should throw my hat into the ring about the controversy surrounding the
ALLHAT trial. A trial which wins my official tortuous acronym award? ALLHAT
stands for the Antihypertensive and Lipid-Lowering
treatment to prevent Heart Attack Trial. Actually,
my favourite acronym is the CARPORT trial. This stands for, wait for it: the
Coronary Artery Restenosis Prevention On Repeated
Thomboxane A2-receptor blockade. There are others, the
TIBET, ALIVE, LIFE, LIMIT. Rule number one, your clinical trial must have a
memorable acronym otherwise, self-evidently, no-one will remember it. Anyway,
what is the ALLHAT trial, and what has it shown? The first thing to note is
that it is a big trial, with more than 40,000 patients in it. And it is also
a long trial; the followup was designed to last at least 6 years. So the
results carry a little more weight than the old ‘five patient, twenty
eight day study in Tibetan Yak herders.’ The
trial had two parts, an antihypertensive part and a lipid lowering part. The
purpose of the trial was to assess the incidence of fatal coronary heart
disease and nonfatal MI in patients treated with chlorthalidone (a diuretic),
amlodipine (a calcium channel blocker), lisonpril (an ACE inhibitor) or
doxazosin (an alpha blocker). In the lipid lowering population the plan was
to assess the all-cause mortality in those treated with either pravastatin
or ‘usual care.’ The lipid lowering study had half the patients in it. In
the hypertensive study, the doctor could start treatment with any of the
four antihypertensive drugs. If that didn’t lower the blood pressure
enough they could increase the dose, then, if that didn’t work, add in the
other agents. One thing of note in this trial is that 55% of patients had to
be black, and 45% women. Mainly because most clinical trials had been done
in white Caucasian males, and there are racial and sex differences in
treatment effects. So
what were the investigators trying to find out? Two
main things. Firstly, is any form of antihypertensive agent better than any
other at preventing fatal CHD and non-fatal MI? Secondly,
does lipid lowering with a statin have any effect on all-cause mortality.
That is, dying of anything. A
couple of questions immediately arise. Why did they use the terminology
‘fatal CHD and non-fatal MI’ in the hypertensive part of the trial? If
fatal CHD is not fatal MI, then what is it? Also, why did they choose to
look at different end-points for the statin, namely all-cause mortality? Why
didn’t they choose to look at all cause mortality in the hypertensives? Trials
are always much more interesting, in my opinion, for the questions they
choose not to ask, than for the questions they actually ask. And,
generally, the questions that the trial didn’t ask form the first point of
attack on any trial — that doesn’t provide the results that people agree
with. ‘Oh, well, what do you expect from ALLHAT, they didn’t look at
fatal MI. Which is really important. Frankly, therefore the entire trial is
a complete waste of time….. Pass the port old boy.’ In reality no trial
can ever look at all end points. It’s just not logistically possible. And
if the nit-pickers decide to get to work, any trial can be pulled to bits.
This is exactly what is happening to the ALLHAT trial. Why
is it being pulled to bits? Because
it quite clearly did not show what people wanted it to show. Firstly,
it showed that any form of blood pressure lowering tablet had pretty much
the same effect as any other. (This trial did NOT measure the effect of
placebo, so all results are one tablet verses another). In short, a diuretic
was just as good as a calcium channel blocker, or an ACE-inhibitor. (Alpha
blockers got pulled out of the trial early on as they showed an alarming
increase in death rates). So
what, you might think. No surprise there. Why the attack? Well,
if I were tell you that diuretics are very cheap, have been around for years
and years, and no pharmaceutical company makes any money from selling them;
whereas ACE inhibitors and calcium channel blockers are much more expensive,
and sell billions of dollars worth ever year….. Then you might, just might,
feel that the answer could be found in this area. Personally,
I always ask the question: Is the person attacking the ALLHAT study making
money from companies that make ACE-inhibitors or calcium channel blockers?
If not, I take the comments very seriously. However, I have yet to identify
anyone attacking the hypertensive arm of the ALLHAT study who has zero
financial connections — although they can sometimes appear very tenuous. And
what of the lipid lowering part of the trial? What did that show? Well,
slightly to my surprise…not. The impact of pravastatin on overall
mortality was absolutely zero. Actually, it wasn’t exactly zero; the
overall mortality was slightly higher in the pravastatin group than the
group taking placebo. How
is this being explained? "Both
the pravastatin and usual care groups had substantial cholesterol reductions,"
said Whelton. "This is probably because many of those in the usual care
group received a cholesterol-lowering drug. The magnitude of the trend
toward increasing use of cholesterol-lowering drugs in usual care during the
8 years of the trial reflects the impact on clinical practice of the many
positive statin trials that have taken place in those years. This trend was
not fully anticipated when ALLHAT began in 1994. Thus, no difference was
found between the groups in deaths and only a modest difference in the rates
of heart attack and stroke." That
quote taken from NIH website http://www.nih.gov/news/pr/dec2002/nhlbi-17.htm
For
those of us who enjoy the use of weasel words and non-scientific rubbish
dressed up as fact, that is a paragraph to savour. Examine the sentence that
begins… ‘This is probably because….’ A perfect statement. Not
based on any facts, or data, just a guess. The paragraph then goes on to
state that if this guess is correct, then it explains why statins didn’t
prevent a single death. (Despite the fact, of course, that LDL levels were
lowered much more in the Pravastatin group). So,
all the results of a major clinical trial can be demolished by an unproven
supposition based on no facts whatsoever. Yes folks, it’s science at its
very best. ‘Ignore the results from the ALLHAT trial, it’s rubbish.
Things that we never bothered to measure probably caused the negative
results.’ Can
you imagine what would happen if I said that I just ‘guessed’ that in
other lipid lowering trials, people probably stopped smoking in the statin
group and not in the placebo group, which explains any beneficial effects
seen. Of course I didn’t bother measuring this, I just kind of guessed
that it must be so. How else could you explain the benefits of statins? I must stop now before my blood pressure gets too high and I have to start taking an ACE-inhibitor. I also seem to have got dragged off blood pressure and onto statins. I must be obsessed.
THE
JOY OF HYPERTENSION TRIALS
Yes,
I know, the title looks a bit like one of those horrible mathematical proofs
that only the class swot used to know. Everyone else doodled on the side of
the page and waited for the class bell to ring in time for them to make
their escape. But
please don’t run and hide, I intend to use no mathematics in this article.
What I want to highlight is the fact that a great deal of research directly
contradicts the conventional and yet no-one in the medical community appears
even slightly concerned. Instead,
what happens is that, when fact A contradicts fact B, those who like fact A
will quote it as a reference to support their work. Whereas, those who
prefer fact B will quote that instead. In
order to help clarify an issue that may seem rather abstract, I will provide
a concrete example. Last year two researchers called Law and Wald wrote a
paper in the British Medical Journal which suggested that there is no level
of cholesterol that should not be lowered further. The lower the better,
there is no lower limit, and no age limit. I
objected to such obvious nonsense in a letter to the BMJ. Even if you do
believe that a high cholesterol level causes coronary heart disease (CHD),
you surely cannot also believe that a low cholesterol level causes CHD. If
that’s true then: In
the middle of the night Anyway,
I quoted a major study done in Honolulu demonstrating quite clearly that, in
the elderly, a low cholesterol level was the single most important risk
factor for all cause mortality, and death from CHD. On this basis how could
Law and Wald support lowering cholesterol in the elderly population? On
that specific point Law and Wald stated that it had been ‘proved’ that a
low cholesterol level was the sign of an underlying disease, and it was the
underlying disease that killed people, not the low cholesterol level. They
quoted a paper by Iribarren in support of this concept. However,
the paper itself was just a hypothesis paper, unsupported by any data.
Despite this, the hypothesis is widely accepted to be true. Indeed, the
Honolulu researchers were acutely aware of that hypothesis paper, and had
chosen to analyse this issue closely. In the end, they could find no sign
whatsoever that low cholesterol was caused by any underlying disease. To
quote from that paper, which can be found in the Aug 2001 issue of the
Lancet: ‘Iribarren
and colleagues suggested that a decline in serum cholesterol might occur
over a decade before diagnosis of disease, and such long-term morbidity
could be attributable to chronic subclinical infections with hepatitis B, or
to chronic respiratory disease resulting in repeated respiratory infections.
These disorders could increase concentration of pro-inflammatory cytokines
that cause hypocholsterolaemia. Our present analysis suggest that this
hypothesis is implausible and is unlikely to account for the adverse effects
of low cholesterol over twenty years.’ Which
is the language that one set of researchers use to tell another set of
researchers that they are talking bollocks. As
far as Law and Wald are concerned, however, I’m sure they still firmly
believe that a low cholesterol is caused by an underlying condition, and
that a low cholesterol level is still a good thing. They have no proof of
this; they just like it as a hypothesis. But either this is right or it is
wrong. And it is also an extremely important point. Shouldn’t someone be
trying to establish the truth one way or another? All
of this highlights the fact that utterly contradictory papers can co-exist
in the medical research universe, and no-one ever challenges them to fight
it out mano a mano. Everyone just carries on ploughing their own little
furrows, never daring to peek over the edge. Which,
in a very roundabout way, brings me back to hypertension. Here
are three facts that currently co-exist in the world of medical research.
Starting in the early nineteen eighties when the Medical Research Council (MRC)
UK carried out the first ever long-term study into the effect of blood
pressure lowering on mortality and morbidity. The drugs used were a diuretic
and a beta-blocker. Up
to this point, you may be surprised to hear, this issue had never been
studied. It was sort of assumed that a high blood pressure caused CHD, so if
you lowered the blood pressure, you would prevent CHD. The trial was set up
to provide glorious vindication of this hypothesis Unfortunately
"glorious," and "vindication," are not quite the words
that we should use for this trial. For the primary finding of the MRC trial
was that blood pressure lowering had no impact on the rate of death from CHD.
(There was some reduction in stroke and renal failure.) Jumping
to the present day, the ALLHAT study recently showed that there was no
difference in CHD prevention between diuretics, beta-blockers,
ACE-inhibitors and Calcium Channel blockers. However,
a recent meta-analysis in the NEJM
shows that ACE inhibitors do
provide protection against CHD. This
provides an interesting sequence of statements: Placebo
= Beta-blockers and diuretics (in preventing CHD. MRC trial) Beta-blockers
and diuretics = ACE-inhibitors and calcium channel blockers (ALLHAT) ACE-inhibitors
are > placebo (NEJM) Put
more simplistically a:
A = B Now,
it almost goes without saying that it is impossible for all three statements
to be true. One of them must be false. The question is, which one? Surely
this is of high importance? I believe it is, yet the medical research
community seems perfectly willing to accept all three statements and carry
on regardless. In
my view, if no-one is willing to debate these issues out in the open and
make some kind of decision, then the whole area is a complete mess. And this
is typical of the whole area of research into heart disease. And how can the
public know what to believe when we have data out there that is utterly
contradictory? This
is the reason why you get headline after headline stating that, for example
‘Coffee protects against heart disease,’ followed by ‘Coffee causes
heart disease,’ followed by ‘Coffee protects against heart disease….
Ad infinitum. Well, either coffee does, or does not cause heart disease. Surely
the editors of medical journals should try to create some kind of consensus
on such matters. But they don’t, and so we all sail onto into more and
more confused waters. Perhaps I should try to start a new journal. The
Journal of Medical Logic and Argument — or something of the sort. Where
researchers are forced to debate all areas where there is directly
contradictory evidence. Some hope. Just to sign off. If you want to know my opinion on which statement is false it’s C > A. You see I’m a democrat, and statements A and B support each other, whereas statement C contradicts the others. So we have a two to one majority against C.
IDIOTIC
THINKING IN MEDICINE
You
may have heard a bit about a substance in the blood called C-reactive
protein (CRP). It is released in ‘inflammatory’ conditions in the body:
infections, rheumatoid arthritis and also heart attacks. CRP has been around,
and known about, for years. However, CRP is about to suffer the same fate of
other innocent substances in the blood that have the misfortune to rise in
people who have heart disease. It is going to be accused of causing heart
disease. It
is a dispiriting fact that, when faced with diseases of unknown cause, the
medical profession unerringly manages to get cause and effect completely the
wrong way round. In the world of heart disease this has happened with blood
pressure, LDL, visceral obesity, insulin resistance and HDL - to name but
five. Now it is going to happen to CRP. When
you find an abnormality of some sort that is associated with a disease, you
can make a number of different conjectures: 1.
The abnormality is caused by the disease 2.
An underlying problem causes both the
abnormality and the ‘disease’ 3.
The disease is caused by the abnormality 4.
It’s a coincidence (one in twenty chance) 5.
You haven’t measured things properly You
would think it wouldn’t be that difficult to sort things out, but if you
get things the wrong way round to start with, it can takes years, decades -
forever? Take high blood pressure. In some cases there is a clear underlying
cause e.g. renal artery stenosis. But in the vast majority of cases there is
no (clearly) definable cause. At
this point, rather than say ‘you have high blood pressure of unknown
cause,’ the medical profession decided to use a bit of jargon, and so the
term ‘essential hypertension’ was born. It means exactly the same thing,
but it sounds more scientific and impressive. Twas
but a small step from here to suggest that essential hypertension wasn’t
just a sign of some underlying abnormality; it was, in fact, a disease. A
disease that needed to be treated. And because essential hypertension was
found to be associated with heart disease, it was further decided that if
you ‘treat’ hypertension, then you would ‘cure’ heart disease. Despite
this, many of you probably still think that treating essential hypertension
does reduce the risk of heart disease. But, of course, it doesn’t. A fact
so carefully hidden behind all sorts of barriers that it can take months to
work it out. Finally, you realise that when you read ‘reduction in CV
events’ this doesn’t mean reduced rate of death from Coronary Heart
Disease (CHD). It primarily means reduced rate of death from stroke. Hey
guys, I can see how a high blood pressure might burst the arteries in your
brain. But I can’t see how high blood pressure causes the build up of
plaques in the arteries. On the other hand I can see how narrowing an artery
with a plaque might reduce blood flow, and trigger a response by the body to
raise the blood pressure to keep the blood flow up. Cause that’s simple
fluid dynamics. In short, CHD (or atherosclerotic plaques) causes a high
blood pressure - AND NOT THE OTHER WAY ROUND. The
example of high blood pressure serves as a lesson in how to turn an
‘associated symptom’ into a disease, and how to get cause and effect
hopelessly mixed up. With CRP, we are going to see exactly the same thing.
History is in the making, right now, in front of your very eyes. As
a raised CRP is now a recognised risk factor for heart disease, t’will be
a very small step to suggest that by lowering it, you will prevent heart
disease. I can already see the CRP lowering agents being lined up by the
pharmaceutical companies. Watch for the buzz words IL-6 and
hyper(c)-proteinaemia. You read it here first. A word of warning. Whisper it quietly ‘c-reactive protein reducing agents won’t work.’ |